
Abstract
Protein self-association is a widespread phenomenon that results in the formation of multimeric protein structures with critical roles in cellular processes. Protein self-association can lead to finite protein complexes or open-ended, and potentially, infinite structures. This review explores the concept of protein agglomeration, a process that results from the infinite self-assembly of folded proteins. We highlight its differences from other better-described processes with similar macroscopic features, such as aggregation and liquid-liquid phase separation. We review the sequence, structural, and biophysical factors influencing protein agglomeration. Lastly, we briefly discuss the implications of agglomeration in evolution, disease, and aging. Overall, this review highlights the need to study protein agglomeration for a better understanding of cellular processes.
Similar content being viewed by others


Introduction
Protein self-association is a prevalent phenomenon across organisms (Goodsell and Olson 2000; Levy and Teichmann 2013), primarily driven by non-covalent interactions. Such associations give rise to multimeric protein structures that play crucial roles in diverse cellular processes, including signal transduction (Park et al. 1999), allosteric regulation (Monod et al. 1965), and protein homeostasis (Sigler et al. 1998).
These associations can take finite forms, resulting in closed assemblies (Ahnert et al. 2015; Levy et al. 2008), or infinite forms, leading to the formation of open-ended, often insoluble supramolecular structures (Dobson 2003; Garcia-Seisdedos et al. 2019; Yeates 2017). Traditionally, apart from cytoskeleton proteins, like actin, tubulin, and intermediate filaments (Alberts et al. 2002), infinite assemblies have been linked to misfolding and aggregation (Garcia-Seisdedos et al. 2019). When proteins misfold, hydrophobic residues become exposed to the solvent, leading to non-specific interactions with other misfolded proteins, resulting in the formation of amorphous (Boatz et al. 2017) or amyloid-like aggregates (Dobson 2003). Protein aggregation is typically irreversible, toxic to cells, and frequently associated with diseases (Chiti and Dobson 2006; Dobson 2003; Fink 1998).
However, recent research challenges the notion that infinite assemblies necessarily arise from misfolding. Point mutations, as observed in hemoglobin S in sickle cell disease (Eaton and Hofrichter 1990; Harrington et al. 1997) and certain mutant forms of γD-crystallin in cataracts (Boatz et al. 2017), can induce folded proteins to self-assemble into infinite assemblies (Empereur-Mot et al. 2019; Garcia-Seisdedos et al. 2017; Garcia Seisdedos et al. 2022). Changes in the intracellular environment can also trigger metabolic enzymes to assemble into filaments (Munder et al. 2016; Narayanaswamy et al. 2009; Park and Horton 2019; Petrovska et al. 2014). These assemblies appear to serve various functions, such as regulating enzymatic activity (Kim et al. 2010; Stoddard et al. 2020), preventing degradation (Petrovska et al. 2014), promoting cellular dormancy (Montrose et al. 2020), and, in some cases, being associated with aging (Paukštytė et al. 2023).
Crucially, in contrast to protein aggregates, these cases share the common characteristics of maintaining folded protein structures within the assemblies and displaying reversibility (Garcia-Seisdedos et al. 2019). Thus, to distinguish this phenomenon from aggregation, we propose the term “agglomeration” (Garcia-Seisdedos et al. 2019).
In this review, we define the term agglomeration and highlight its differences from other better-described processes with similar macroscopic features, such as aggregation and liquid-liquid phase separation. We review the structural and biophysical factors influencing protein agglomeration and briefly give an overview of the implications of agglomeration in evolution and disease.
What is protein agglomeration?
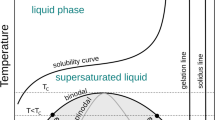
In vitro, protein precipitation is familiar to every molecular biologist with some experience in protein purification. Under certain conditions, such as mutations or changes in buffer composition, proteins can become insoluble, which can complicate subsequent biochemical studies. Protein precipitation has traditionally been attributed to misfolding and aggregation, a process driven by a loss of protein stability (Chiti and Dobson 2017). At the same time, techniques like salt-driven precipitation, using agents such as ammonium sulfate, have long been employed in protein purification (Duong-Ly and Gabelli 2014; Wingfield 2001). It is generally accepted that these techniques do not induce protein misfolding; instead, they reduce the solubility of folded proteins, which can be easily resolubilized using standard buffers (Duong-Ly and Gabelli 2014; Wingfield 2001). These seemingly contradictory phenomena underscore the diverse origins of protein insolubility, challenging the default association of this process with aggregation.
In vivo, i.e., at the cellular level, the widespread use of fluorescent protein tags and advances in fluorescent and electron microscopy techniques, have enabled the visualization and characterization of various biomolecular condensates. These structures include membrane-less bodies like stress granules (Cherkasov et al. 2013; Wallace et al. 2015), germ granules (Brangwynne et al. 2009), and p-bodies (Decker and Parker 2012), as well as protein filaments (Alberti et al. 2009; Petrovska et al. 2014; Stoddard et al. 2020).
Although protein precipitates and biomolecular condensates share similar in vitro and in vivo macroscopic features, they result from fundamentally different biophysical processes, such as aggregation, liquid-liquid phase separation (LLPS), and agglomeration. While protein aggregation and LLPS are well-described processes that rationalize how proteins can demix from the solvent in different liquid or solid phases (Hyman et al. 2014; Sontag et al. 2014), agglomeration has received far less attention and has often been confused with aggregation, emphasizing the need to differentiate these phenomena for a comprehensive understanding of their cellular functions.
Agglomeration
We define agglomeration as the infinite assembly of folded proteins (Garcia-Seisdedos et al. 2019). Agglomerates exhibit several distinctive features: (i) In agglomeration, the constituents of the assemblies, referred to as protomers, are folded proteins. Therefore, in agglomeration, protomers acquire new intermolecular contacts, driving their self-assembly. Importantly, they retain all of their native contacts and maintain their native structure. (ii) Agglomerates are open-ended and potentially infinite assemblies. This means that as long as the concentration of free protomers is sufficiently high, agglomerates can continue to grow in size. In some cases, they can form micron-scale protein structures within cells (Garcia-Seisdedos et al. 2017; Garcia Seisdedos et al. 2022). (iii) Agglomeration is a reversible process. Meaning that protomers within the assemblies can dissociate from them. This dissociation can occur due to conformational changes (Eaton and Hofrichter 1990), dilution (Garcia-Seisdedos et al. 2017), or environmental changes, such as variations in pH or macromolecular crowding (Munder et al. 2016; Petrovska et al. 2014). Importantly, this dissociation happens without the need for a third protein partner, such as disaggregases or other components of the proteostatic machinery.
Differences between agglomeration and aggregation
Unlike agglomeration, aggregation is driven by misfolding (Chiti and Dobson 2006). Protein constituents of aggregates lose part of their native contacts which are replaced by new ones with other misfolded proteins, resulting in their assembly into high-order structures (Fig. 1). Aggregates, like agglomerates, are also potentially infinite structures. However, unlike agglomerates, aggregates are not reversible. They are often low-energy hyper-stable structures requiring energy-consuming cellular components like disaggregases and other chaperones (Bukau et al. 2006; Rosenzweig et al. 2019) for dissociation. There are some exceptions though, as some protein aggregates have been observed to dissolve and refold upon dilution (Iadanza et al. 2018). However, the extremely slow kinetics of dissolution and refolding make this process generally irrelevant to biological timescales (Iadanza et al. 2018). Lastly, while aggregation is generally deleterious for fitness (Geiler-Samerotte et al. 2011), agglomeration does not necessarily pose a threat to the cell, as supported by the physiological roles of many agglomerates formed by natural proteins (Nüske et al. 2020; Petrovska et al. 2014; Prouteau et al. 2017; Stoddard et al. 2020). It is worth mentioning that prions constitute a special group of aggregates. Prions are misfolded proteins that self-propagate of their folded counterparts. Although functional prions are common, and whether they are misfolded or low-complexity protein conformations are debatable, they are usually prevented by the presence of high kinetic barriers (Franzmann and Alberti 2019)

Contraposition of agglomeration and aggregation concepts. Aggregation is mediated by protein misfolding, which can be driven by destabilizing mutations or by changes in the environment (e.g., temperature, pH). Misfolded proteins can assemble into ordered structures such as amyloid beta or amorphous aggregates such as the ovotransferrin aggregates (images reproduced from Gremer et al. (2017) and Constantinescu et al. (2017)). In contrast, agglomeration is mediated by the creation of a new protein interaction between two well-folded proteins. Such interaction can be induced by a mutation, as in sickle cell disease, or by changes in the environment. Agglomerates can be ordered, as the filaments of hemoglobin s in sickle cell disease, or amorphous, as in some types of cataracts (images reproduced from Harrington et al. (1997) and (Boatz et al. 2017))
While agglomeration and aggregation are distinct, instances like domain swapping blur their boundaries. In this scenario, supramolecular assemblies are composed of interlaced well-folded protomers that retain most of their native contacts (Guo and Eisenberg 2006). Some examples of domain swapping are RNase A (Guo and Eisenberg 2006; López-Alonso et al. 2010) and bacterial pili (Busch and Waksman 2012). However, the unfolding of the protomers precedes domain swapping (López-Alonso et al. 2010). In that respect, we could consider domain swapping midway between aggregation and agglomeration.
Differences between agglomeration and LLPS-driven condensates
A widespread class of membrane-less bodies consists of condensates formed through liquid-liquid phase separation (LLPS). These LLPS condensates exhibit similarities with agglomerates, as both structures involve native proteins, are reversible, and form open assemblies (Alberti and Hyman 2021). However, there are notable differences between these two processes: (i) LLPS is often mediated or associated with intrinsically disordered domains (Banani et al. 2017; Heidenreich et al. 2020; Pak et al. 2016; Wright and Dyson 2015), resulting in liquid-like properties (Banani et al. 2017; Gomes and Shorter 2019; Hyman et al. 2014). In contrast, agglomerates consist of folded protein units that assemble into either disordered (Boatz et al. 2017; Pande et al. 2005) or ordered solid-like structures, such as filaments (Garcia-Seisdedos et al. 2017; Marini et al. 2020), lattices (Gonen et al. 2015), and crystals (Lanci et al. 2012). (ii) Agglomerates are considered homogeneous structures in which the identity of the constituent protomers is well-defined (Garcia-Seisdedos et al. 2019). On the other hand, LLPS condensates are highly heterogeneous in composition, often containing multiple protein types and nucleic acids (Banani et al. 2017).
Factors linked to agglomeration
Protein solubility is defined as the concentration of protein in a saturated solution that is in equilibrium with a solid phase under specific conditions (Arakawa and Timasheff 1985; Kramer et al. 2012; Scatchard et al. 1943). Solubility is influenced by a range of factors, including both intrinsic and extrinsic elements.
In the context of agglomeration, intrinsic factors encompass aspects such as the symmetry of the quaternary protein structure and the surface chemistry of the protein (Empereur-Mot et al. 2019; Garcia-Seisdedos et al. 2017, 2019; Garcia Seisdedos et al. 2022). Extrinsic factors, when considering cellular processes, include variables like intracellular protein concentration and the physicochemical properties of the cellular environment (Munder et al. 2016; Petrovska et al. 2014). In this section, we will analyze these factors.
Symmetry of quaternary structure
Agglomeration of proteins is intimately tied to the symmetry of their quaternary structure. Symmetric complexes are prevalent in agglomerates, and specifically, dihedral homomers represent more than 60% of proteins known to form agglomerates (Garcia-Seisdedos et al. 2019). Dihedral homomers are those protein complexes that form in such a way that they exhibit two orthogonal axes of rotational symmetry, with one of them being twofold. This implies that the complex can be rotated 180° around one of the axes and still maintain its original configuration. The overrepresentation of dihedral homomers in agglomerates likely responds to several factors:
-
1-
Structural and sequence similarity increases the interaction propensity of proteins (Lukatsky et al. 2007; Wright et al. 2005). Partly because the probability of finding a self-matching pattern between two identical surfaces is higher compared with the probability of a pattern match between two different surfaces (Lukatsky et al. 2007). Additionally, when two identical surfaces interact, each amino acid residue is repeated twice, increasing its contribution — either favorable or unfavorable — to the free energy association (Lukatsky et al. 2006; Wales 1998). Thus, in a context where interfaces are created randomly and stable ones are selected, interfaces involving two identical protein surfaces have higher chances of selection than those composed of distinct ones (André et al. 2008; Lukatsky et al. 2007; Schulz 2010). This phenomenon could explain the anomalously higher fraction of homodimers relative to heterodimers observed across proteomes (Ispolatov et al. 2005).
-
2-
The repetition of subunits within symmetric complexes introduces multivalence. This property is central in polymer and supramolecular chemistry. Multivalent molecules naturally assemble into polymers or oligomers when mixed, decreasing molecules’ solubility due to entropy-driven effects (Flory 1953), which might promote their phase separation (Banani et al. 2017). Multivalency has been harnessed for the design of distinct protein agglomerates such as protein fibers (Garcia-Seisdedos et al. 2017; Garcia Seisdedos et al. 2022; Grueninger et al. 2008; Padilla et al. 2001), lattices (Gonen et al. 2015; Padilla et al. 2001; Suzuki et al. 2016), and crystals (Lanci et al. 2012; Padilla et al. 2001). Furthermore, the interaction between two symmetric complexes potentially implies multiple binding sites, which increases the strength and specificity of interactions in a cooperative manner (Curk et al. 2017; Fasting et al. 2012; Romero Romero et al. 2018).
-
3-
New self-interactions among symmetric complexes often result in open-ended assemblies (Empereur-Mot et al. 2019; Garcia-Seisdedos et al. 2017, 2019; Yeates 2017). When a new self-interaction is created on a monomer or a cyclic complex, it will likely result in a dimer or a finite complex. Whereas when occurring on a complex with dihedral symmetry, the new self-interaction will necessarily trigger an open-ended, and potentially infinite, assembly (Fig. 2a).

Intrinsic factors linked to agglomeration. a Symmetry of protein structure is intimately tied to agglomeration as it confers multivalence. A monomeric protein gaining a self-interacting patch forms a dimer, whereas a self-interacting patch repeated on opposite sides of a symmetric protein may result in an infinite assembly. b On average, only two substitutions are required to shift the average composition of a surface patch into an interface patch and vice versa (Levy 2010)
Protein surface chemistry
For a new protein assembly to take place, a new protein interaction must be created. Theoretical work showed that the chemical composition of a protein interface is very similar to the protein surface. In fact, only two amino-acid substitutions are enough to turn the average surface patch into the average interface patch in terms of chemical composition (Fig. 2b) (Levy 2010). This work suggests that point mutations often lead to the formation of new protein assemblies. If these assemblies are open-ended, they can potentially give rise to new agglomerates.
Several experimental works support this theory. In sickle cell disease, a point mutation at the surface of human hemoglobin that substitutes a charged residue with a hydrophobic one (mutation E6V) results in the formation of a new interface between hemoglobins. As human hemoglobin is symmetric, the acquired interaction results in the formation of fibers (Eaton and Hofrichter 1990). In recent work, Garcia-Seisdedos et al. examined whether point mutations designed to increase the hydrophobicity of the surface of 12 dihedral homomers would trigger similar results. They showed that all 12 homomers underwent agglomeration, confirming that minimal increments in the hydrophobicity of proteins’ surfaces frequently drive new self-interactions (Garcia-Seisdedos et al. 2017).
In a different work, the same authors performed random mutations at the surface of two dihedral homomers. The results indicated that two different mutational pathways lead the homomers to agglomerate. One route involved increasing proteins’ surface hydrophobicity, as we previously demonstrated (Garcia-Seisdedos et al. 2017). A second route implies the sole removal of charged residues from the surface. Notably, replacing a few charged residues for alanines in eight dihedral homomers confirmed that the sole neutralization of surfaces commonly drives agglomeration and intracellular changes in the localization of proteins (Garcia Seisdedos et al. 2022).
The burial of hydrophobic residues at protein interfaces is energetically favorable due to desolvation effects (Chothia and Janin 1975; Garcia-Seisdedos et al. 2012; Romero et al. 2022). It is thus expected that mutations introducing hydrophobic residues are prone to trigger new self-interactions. Indeed, hydrophobicity in protein surfaces can be a burden that is often counterbalanced by negative design, where specific chemical properties evolve to minimize the risk of undesirable protein association events (Doye et al. 2004). Taking into account that protein interactions are concentration-dependent, highly expressed proteins are at particular risk, and consistently, they generally exhibit less hydrophobic interfaces to prevent promiscuous interactions (Levy et al. 2012). Negative design can operate not only on specific proteins but also on sensitive sites on their surface to reduce undesirable self-assembly events (Empereur-Mot et al. 2019; Garcia-Seisdedos et al. 2017; Pechmann et al. 2009)
Even more surprising is that simply removing charges from protein surfaces can create new interfaces (Garcia Seisdedos et al. 2022). This implies the presence of interaction-prone patches on protein surfaces, which are normally shielded by charged residues to prevent their association. Why such sticky patches exist in some protein surfaces is not fully understood. One possibility is that these sticky patches might facilitate the conditional assembly of proteins under specific cellular environments, such as changes in pH (Garcia Seisdedos et al. 2022). A decrease in pH could neutralize gatekeeper charges on protein surfaces, thereby enabling protein self-association. Such a phenomenon is suggested to regulate some enzymes’ oligomerization (Andréll et al. 2009) and could plausibly explain the pH-dependent agglomeration of many metabolic enzymes (Munder et al. 2016).
Disulfide bridges between cysteines and metal coordination can also trigger intermolecular association of protomers and promote agglomeration in vitro (Suzuki et al. 2016). In this work, Suzuki and co-workers minimally redesigned the surface of a C4 homomer by introducing cysteines and histidines. Thus, oxidation of the cysteines and coordination of Zn2+ and Cu2+ ions by histidines resulted in the formation of two-dimensional lattices. A similar strategy was followed to create one-, two-, and three-dimensional protein arrays through the coordination of Zn2+ ions also by means of surface histidine residues (Brodin et al. 2012). Previously, Lawson and co-workers designed a ferritin crystal by coordination of Ca2+ ions through glutamine and aspartic residues (Lawson et al. 1991). In addition to proteinogenic amino acids such as histidines, glutamines, aspartic, and glutamates (Song et al. 2014), metal coordination can also be achieved by non-natural and chemically modified amino acids in the design of protein agglomerates (Tavenor et al. 2017; Yang and Song 2019).
Other kinds of interactions, such as Met S-Aromatics, have the potential to create protein agglomerates (Valley et al. 2012).
Protein concentration
Protein binding is a process that depends on the concentration of its constituents. In principle, every protein could agglomerate provided that its concentration is high enough. Consequently, proteins that are highly expressed in the cells are at particular risk of protein agglomeration (Fig. 3a). Indeed, sickle cell disease happens as a result of a mutation in human hemoglobin, whose intracellular concentration in human red blood cells is exceptionally high — on the order of 330 mg/mL (Krueger and Nossal 1988). Similarly, some types of cataracts are associated with mutations in γS-crystallin, whose concentration in the eye lens is higher than 400 mg/mL (Brubaker et al. 2011). Consistently, highly expressed proteins evolve slowly (Yang et al. 2012), and their surfaces are subjected to negative design — show less sticky surfaces — (Levy et al. 2012) presumably to avoid misinteractions.

Extrinsic factors linked to agglomeration. Protein concentration, pH, macromolecular crowding, viscosity, and co-solutes are extrinsic factors modulating protein agglomeration
The risk of agglomeration is not restricted to highly expressed proteins, though. On the one hand, point mutations, especially when occurring in oligomers, can drive protein self-assembly at low concentrations (Garcia-Seisdedos et al. 2017). On the other hand, proteins have different self-assembly potentials (Garcia Seisdedos et al. 2022), due to factors such as symmetry and surface chemistry, and so, the concentration threshold at which different proteins agglomerate is different. While insolubility is suggested to shape the expression levels of proteins in the cell (Tartaglia et al. 2007), the intracellular concentration of most proteins seems to be close to or even above their solubility threshold (Tartaglia et al. 2007; Vecchi et al. 2020). Therefore, we could expect agglomeration events to be common in proteomes.
Cellular environment
As cells are dynamic entities, so are the biophysical properties of their interiors, which cells often regulate or harness to adapt to changing environments (Garcia-Seisdedos et al. 2020; Villegas et al. 2022). For example, stress and nutrient depletion are often met with massive intracellular agglomeration events and reorganization of molecules (Minsky et al. 2002; Munder et al. 2016). Such changes in the assembly state of molecules might be provoked by changes in pH (Munder et al. 2016; Petrovska et al. 2014), macromolecular crowding (Delarue et al. 2018), solvent properties (Patel et al. 2017), and viscosity (Persson et al. 2020). Here, we comment on how these biophysical properties can affect the agglomeration of proteins (Fig. 3b–e).
Changes in pH
The effects of pH on protein solubility have been known for decades. In his book, Physical Chemistry of Macromolecules, Charles Tanford noted that to a first approximation, protein solubility is proportional to the square of its net charge (Tanford 1961). Thus, when the pH of a solution comes near the isoelectric point of a protein — the pH value at which the net charge is zero — the solubility of a protein drops to its minimum (Tanford 1961). Under these conditions, proteins display low net charge and are subject to weaker repulsive interactions, making attractive interactions dominant (Fig. 3b). Therefore, provided that their concentration is high enough, proteins can self-assemble into agglomerates (Munder et al. 2016).
While the effect the pH on the in vitro agglomeration of proteins was known for decades (Boye et al. 1996; Matsudomi et al. 1991; Parker et al. 2005; Renard and Lefebvre 1992), only some decades later was reported the influence of pH on protein agglomeration in a cellular context (Munder et al. 2016; Nüske et al. 2020; Petrovska et al. 2014), possibly due to the erroneous assumption that cytosolic pH was invariable in cells (Petrovska et al. 2014). Petrovska and co-workers showed that the enzyme glutamine synthase forms filaments in yeast upon acidification of the cytosol (Petrovska et al. 2014). Later, the same research group found that low pH promoted a general liquid-to-solid transition of the yeast cytosol that includes the agglomeration of many metabolic enzymes (Munder et al. 2016) and translation factors (Nüske et al. 2020). Notably, changes in pH also trigger the formation of stress granules through liquid-liquid phase separation in yeast (Riback et al. 2017).
A drop in the intracellular pH may trigger the neutralization of charges of glutamic and aspartic residues of proteins’ surfaces, decreasing the repulsive interactions and promoting the agglomeration of some proteins. Indeed, the neutralization of a few charged residues is sufficient to trigger agglomeration in many proteins (Garcia Seisdedos et al. 2022). As intracellular acidification is common upon energy depletion and other stress conditions (Orij et al. 2012; Pintsch et al. 2001; Riback et al. 2017; Weitzel et al. 1985), and negatively charged proteins with acidic pI dominate in the cytosol across organisms (Schwartz et al. 2001), acidic-driven assembly of proteins may be a general adaptive mechanism to stress (Munder et al. 2016).
Macromolecular crowding
In starved cells, macromolecular motility is severely restricted (Joyner et al. 2016; Parry et al. 2014). Joyner and co-workers found that such low motility could not be explained by pH changes or low energy levels but rather by increased macromolecular crowding due to water loss and cell size reduction (Joyner et al. 2016). In addition, vacuole enlargement has also been proposed to play a role in the increase of molecular crowding in starved yeast cells (Marini et al. 2020). Macromolecular crowding is also directly regulated by the central growth regulator mTORC1 by tuning the concentration of ribosomes in the cell, through production and autophagy (Delarue et al. 2018). Altogether these studies suggest that cells closely regulate the level of crowding in order to control intracellular diffusion and macromolecular interactions (Joyner et al. 2016).
Macromolecular crowding refers to the effects of adding macromolecules to a solution, as compared to a solution without any macromolecules (Aumiller et al. 2014). It is crucial for the functioning of biological systems, as crowding agents in high concentrations entropically favor the association of molecules, accelerating chemical reactions (Zhou et al. 2008). However, excessive crowding dramatically decreases molecular motility (Miermont et al. 2013; Trappe et al. 2001). The impact of crowding on molecules depends on their molecule size; molecules with large sizes are usually more affected than smaller particles (Zimmerman and Minton 1993).
In the crowded environment of the cell, protein-protein interactions are favored, as the excluded volume around the protein complex is smaller than the excluded volume of each monomeric protein unit (Minton 2000; Ralston 1990). Since agglomerates are large oligomers, crowding is expected to have a major impact on protein agglomeration (Minton 2000; Ross and Minton 1979). Indeed, excluded volume theory predicts that the addition of an inert macromolecule can significantly reduce the solubility of a protein that is in equilibrium with a condensed phase, hence promoting the formation of agglomerates and fibrillar assemblies (Ross and Minton 1979) (Fig. 3c). Such theoretical predictions have been experimentally validated. Notably, the addition of inert proteins to solutions of hemoglobin S substantially promotes and accelerates the formation of hemoglobin fibers (Behe and Englander 1978). The addition of crowding agents also lowers the critical concentration and accelerates the formation of actin and tubulin microtubules (Drenckhahn and Pollard 1986; Herzog and Weber 1978; Lindner and Ralston 1997; Tellam et al. 1983; Woodruff et al. 2017), and fibrin clot formation (Wilf et al. 1985).
More recently, Petrovska and co-workers showed that the yeast enzyme glutamine synthase needs a crowding agent at concentrations that resemble the physiological crowding of the cell to reconstitute in vitro filaments observed in starved cells (Petrovska et al. 2014). In consonance, the same research group showed that, in addition to pH, crowding also influences the assembly of a translation initiation factor that agglomerates in starved yeast cells (Nüske et al. 2020). These studies suggest that both pH and macromolecular crowding have strong effects on protein solubility, and they are known driving forces for the formation of agglomerates.
Viscosity
It was recently shown by Person and co-workers that cells regulate the synthesis of trehalose and glycogen to adjust the intracellular viscosity (Persson et al. 2020). This mechanism seems to aid yeast cells in adapting to temperature and low energy level changes by maintaining invariant diffusion rates at different temperatures. Beyond yeast cells, viscosity is tuned to promote an intracellular “glassy” state with low metabolic activity in bacteria (Parry et al. 2014), is increased in plant spores, and linked to low metabolic mobility and metabolic inactivity (Buitink and Leprince 2004), and intriguingly changes in viscosity have been observed in cancer cells (Guyer and Claus 1942; Rousset et al. 1981; Takahashi et al. 1999; Wu et al. 2019).
The variation of intracellular concentrations of trehalose and glycogen that modulate the cell’s viscosity seems to affect protein solubility and phase separation (Persson et al. 2020). Although the effect of viscosity on protein agglomeration has not been well characterized, Person and co-workers’ results suggest that high levels of glycogen and trehalose could decrease protein solubility. Suggesting that viscosity could likely modulate the agglomeration of proteins (Fig. 3d).
Solvent properties
The burial of hydrophobic patches at the surface of proteins is the main contributor to protein-protein interactions (Chothia and Janin 1975), which leads to the release of water molecules in bulk and an increase in entropy. The entropy gained by water compensates for the entropy lost by protein molecules forming a complex. Electrostatic interactions also contribute to the stability of protein complexes and confer interaction specificity (Chothia and Janin 1975; Derewenda 2004; Doye et al. 2004). They require surface complementarity and incorrect associations are heavily penalized by unfavorable enthalpies due to poor packing and loss of hydrogen bonds made to water (Chothia and Janin 1975). Thus, the presence of solutes that change the solvent structure, like hexanediol, modulate the strength of hydrophobic interactions, as well as the concentration of ions impacts the interactions between charged residues (Villegas et al. 2022).
The exploration of the impact of ions on protein interactions dates back more than a century to the work of Franz Hofmeister (Hofmeister 1888). He conducted studies on the salt effects on protein precipitation, particularly focusing on hen egg white proteins. In this context, protein precipitation is caused by salt-mediated protein agglomeration. He found that the ability of salts to precipitate proteins, known as “salting-out,” depended on the hydration properties of the ions. This led to the development of the Hofmeister series, an empirical ranking of both cations and anions based on their effectiveness in precipitating proteins.
These series are divided into anionic and cationic, with anionic exhibiting a stronger effect (Marcus 2009). The series are CO32− > SO42− > H2PO4− > F− > Cl− > Br− > NO3− > ClO4− > SCN−, and NH4+ > Cs+ > K+ > Na+ > Li+ > Ca2+ > Mg2+ > Zn2+, for anions and cations, respectively. Ions are ranked from kosmotropes, also termed “water structure makers,” with strong hydration causing an increase in protein stability and a decrease in protein solubility (salting-out) to chaotropes or “water structure breakers,” that increase protein solubility (salting-in) with a decrease in conformational stability (Hatefi and Hanstein 1969; Marcus 2009; Zhao et al. 2006) (Fig. 3e). Although the implications of these effects are wide in several scientific areas, we still lack a complete mechanistic understanding (Gibb 2019).
Divalent cations also play a role in modulating protein-protein interactions (Arakawa and Timasheff 1984). Their influence on the surface chemistry of proteins can lead to agglomeration, where these cations act as bridging molecules between protomers (see the “Protein surface chemistry” section). In addition, agglomeration can also be mediated via other surface-binding molecules. Those molecules may serve as bridges between protomers bringing their interaction. Bridging molecules can be DNA (Mou et al. 2015), cycles (Alex et al. 2019), heme (Kitagishi et al. 2007), biotin (Burazerovic et al. 2007; Müller et al. 2011; Petkau-Milroy et al. 2013), sugars (Hoeg-Jensen et al. 2005; Sukegawa et al. 2017), and other small molecules (Słabicki et al. 2020).
Agglomeration in evolution
Creating novel protein interfaces leading to agglomerates might create new protein functionalities (Song and Tezcan 2014) and provide enzymatic regulation opportunities (Hvorecny and Kollman 2023). The agglomerating state might lock proteins in conformations, inactivating (Barry et al. 2014; Petrovska et al. 2014) or promoting enzymatic activities (Lynch et al. 2017), and even switching between activities in moonlighting proteins (Moon et al. 2005a). Furthermore, agglomeration might cause difficult substrate accessibility, which can be harnessed to increase substrate selectivity (Kim et al. 2005), and might boost enzymatic cooperation, facilitating substrate channeling (Chang et al. 2015). We recommend referring to Park and Horton’s review (Park and Horton 2019) for an exhaustive review of protein filamentation. We also recommend referring to recent reviews tackling how agglomeration can regulate enzymatic activity (Garcia-Seisdedos et al. 2019; Hvorecny and Kollman 2023; Liu 2016; Lynch et al. 2020; Montrose et al. 2020). Here, we give a brief overview of how agglomeration regulates the activity of some enzymes and how agglomeration can promote cellular dormancy (Montrose et al. 2020; Munder et al. 2016).
Enzymatic regulation
Protein agglomeration can act as a mechanism of allosteric control, either by stabilizing the active or inactive states of an enzyme or by influencing the affinity for other allosteric effectors (Hvorecny and Kollman 2023). A key advantage of agglomeration in enzymatic regulation is that it is a fast and energy-efficient mechanism compared to transcription, post-translational modifications, and enzymatic degradation.
Agglomeration frequently inhibits enzymatic activity. In the case of E. coli CTP synthase, filamentation stabilizes the inactive conformation of CTP protomers and, as a consequence, inhibits CTP activity (Fig. 4a) (Barry et al. 2014). Yeast Glucokinase 1 (Glk1), a key enzyme that regulates entry into glycolysis, is active as a monomer, and it is typically expressed when glucose levels are low. When glucose levels spike, Glk1 assembles into filaments that inactivate its activity by preventing substrate turnover (Stoddard et al. 2020). Also, in yeast, glutamine synthase (Gln1), an essential metabolic enzyme catalyzing the synthesis of glutamine from glutamate and ammonium, assembles in filaments under starvation conditions inactivating its activity (Fig. 4b) (Petrovska et al. 2014). Agglomeration can also inactivate the function of other proteins, this is the case of the yeast translation initiation factor eIF2B (Gcn3), which forms inactive filaments under starvation downregulating protein translation (Fig. 4b) (Nüske et al. 2020). The target of rapamycin serine/threonine kinase (TOR) has two complexes TORC1 and TORC2 that regulate cell growth and metabolism. Recently, it was reported that under glucose deprivation, yeast TORC1 assembles into helical fibers inhibiting TOR activity (Prouteau et al. 2017).

Agglomeration in evolution. a Model of allosteric regulation for human and E. coli CTPS. Although filament formation is present in both organisms, it has opposite effects on the enzyme activity (adapted with permission of Lynch et al. (2017)). b In S. cerevisiae, the enzyme glutamine synthase (Gln1) and the transcription initiation factor eIF2B (Gcn3) form filaments upon glucose deprivation (adapted from Petrovska et al. 2014, and Nüske et al. 2020). c Hydrogen peroxide–induced polymerization of the enzyme 2-cys peroxiredoxin serves as a functional switch from peroxidase to chaperone activity (adapted with permission from Saccoccia et al. 2012). d TEM image displaying eIF2B filaments in a starved yeast cell (image reproduced from Marini et al. 2020)
Agglomeration can also promote enzymatic activity, as exemplified by the human CTP synthases. Filamentation of CTP synthase 1 stabilizes the active conformation increasing enzymatic activity (Fig. 4a) (Lynch et al. 2017). CTP synthase 2 forms dynamic filaments that can switch between inhibited and active states through cooperative interactions between protomers along the filament. This cooperativity enables rapid adaptation to changes in nucleotide demand in cells based on substrate and allosteric inhibitor balance (Lynch and Kollman 2020). Human glutaminase C polymerization into filaments also promotes its catalytic activity, and its allosteric inhibitor, BPTES, inhibits the enzyme by disrupting the filaments (Ferreira et al. 2013). The binding of allosteric activators to the enzyme glutamate dehydrogenase (GDH) increases its activity and induces the formation of filaments. On the other hand, the binding of inactivators decreases the enzyme’s activity and leads to the dissociation of filaments. This suggests an association between enzymatic activity and the filamentous form of GDH (Fahien et al. 1989; Frieden 1959; Gylfe 1976; Park and Horton 2019).
Other enzymes like acetyl Co-A and IMPDH can assemble into filaments that adopt both active and inactive conformations, shifting from one to the other upon binding substrates and allosteric modulators (Anthony et al. 2017; Beaty and Lane 1983; Hunkeler et al. 2018; Kim et al. 2010; Park and Horton 2019; Simonet et al. 2020).
Agglomeration can also switch activities in multifunctional proteins, that is the case of human 2-Cys peroxiredoxins, where the formation/dissociation of filaments triggers a functional switch between chaperone and peroxidase activities (Fig. 4c) (Moon et al. 2005b)
Cellular dormancy
Recent studies have reported that key metabolic enzymes conditionally assemble into filaments in response to nutrient scarcity (Marini et al. 2020; Minsky et al. 2002; Nüske et al. 2020; Petrovska et al. 2014). Some studies propose that when certain enzymes form filaments, they deactivate their function, leading to a metabolic shutdown and putting cells in a dormant state (Marini et al. 2020; Munder et al. 2016; Nüske et al. 2020; Petrovska et al. 2014). Most of these studies have been conducted in S. cerevisiae, where the enzyme glutamine synthase, the translation initiation factor eIF2B, and the master regulator TORC1 have been demonstrated to downregulate their function through agglomeration triggered by nutrient deprivation (Fig. 4b and d) (Nüske et al. 2020; Petrovska et al. 2014; Prouteau et al. 2017). Notably, upon glucose addition, the assemblies dissolve and cells resume growth (Nüske et al. 2020; Petrovska et al. 2014). Thus, protein agglomeration constitutes an energy-independent and reversible mechanism for cells to shut down their metabolism, reducing costs, and entering in a growth-arrested state allowing them to survive the lack of nutrients.
Under starvation, and consequently, under low energy levels, cell interiors can undergo a liquid-to-solid transition (Minsky et al. 2002; Munder et al. 2016; Parry et al. 2014) where general assembly of proteins is promoted (Minsky et al. 2002). In fact, physical factors known to change with cellular state, like pH, crowding, or energy levels, influence intra-cellular protein assembly (Munder et al. 2016; Petrovska et al. 2014) (see section “Factors linked to agglomeration”) and phase-separation (Franzmann et al. 2018; Villegas et al. 2022). In addition to a mechanism to promote metabolic shutdown, agglomeration of proteins has been proposed to constitute storage depots (Munder et al. 2016) and to prevent damage to vital molecular components under acute stress (Minsky et al. 2002).
Agglomeration in disease
The fact that point mutations can frequently cause proteins to form agglomerates, as it has been recently demonstrated (Garcia-Seisdedos et al. 2017; Garcia Seisdedos et al. 2022), suggests that agglomeration could be related to many disease mutations. Perhaps the most paradigmatic case of agglomeration in disease is that of hemoglobin in sickle cell disease (Eaton and Hofrichter 1990), whereby the mutation E6V at the surface of human hemoglobin triggers the formation of filaments causing the red blood cells to acquire their characteristic sickle shape (Fig. 5a). Notably, the filaments form in the de-oxygenated form of the hemoglobin and dissolve upon re-oxygenation exhibiting the characteristic reversible nature of agglomerates. A less famous disease occurring also in hemoglobin is caused by a mutation in the same residue but to a lysine. The hemoglobin mutant E6K agglomerates form intracellular crystals causing hemolytic anemia (Fig. 5a) (Charache et al. 1967; Vekilov et al. 2002). The assemblies are also reversible, but opposite to in sickle cell disease, they form in the oxygenated form and dissolve upon de-oxygenation.

Agglomeration in disease and aging. a Mutations E6V and E6K in human hemoglobin cause sickle cell disease and hemolytic anemia respectively (images reproduced from Harrington et al. 1997, and from the ASH Image Bank — This image was originally published in ASH Image Bank. Luiz Arthur Leite, PhD; Danilo Retucci; Fabiana Conti, MD; Vinicius de Moraes. Title: Sickle cell anemia in a hemolytic crisis. ASH Image Bank. 2021; Image # 00063634. © the American Society of Hematology.). b Mutations P23T and P36T trigger agglomeration of γ-crystallin in the eye lens causing cataracts (images reproduced from Boatz et al. 2017, and Kmoch et al. 2000). c Ribosome crystals isolated from brain cells of a patient with Pick’s disease (image reproduced from (O’Brien et al. 1980)). d The metabolic enzyme glutamate synthase (gtl1) forms agglomerates in aged yeast cells triggering amino acid accumulation and mitochondrial dysfunction that results in a shorter lifespan compared to non-agglomerating mutants of gtl1 (image modified with permission of Paukštytė et al. 2023)
Other diseases associated with agglomeration include cataracts, where some mutants in γD-crystallin form large assemblies of folded proteins in the eye lens scattering light and causing vision impairment (Fig. 5b) (Boatz et al. 2017; Kmoch et al. 2000; Pande et al. 2005). In addition, agglomeration appears to be linked to some mutant forms of SOD1 in ALS (Garcia-Seisdedos et al. 2019; Pratt et al. 2014). Changes in filament assembly parameters of the enzyme IMPDH induced by mutations are linked to rhinitis pigmentosa (Burrell et al. 2022; Labesse et al. 2013; Thomas et al. 2012). Intriguingly, large crystalline assemblies of ribosomes, known as Hirano bodies, can be found in brain cells of aged senile humans, and are a histological feature in brain tissue of patients afflicted with neurodegenerative diseases like Pick’s disease, Alzheimer’s disease, and Creutzfeld-Jacob’s disease (Fig. 5c) (Minsky et al. 2002; O’Brien et al. 1980).
Agglomeration in aging
Recently, Paukštytė and co-workers found that glutamate synthase (Glt1) polymerizes into agglomerates during aging in S. cerevisiae, causing the breakdown of cellular amino acid homeostasis. Notably, they found that inhibiting Glt1 polymerization by mutating the polymerization interface restored amino acid levels in aged cells, attenuating mitochondrial dysfunction, and promoting lifespan extension (Fig. 5d) (Paukštytė et al. 2023).
Concluding remarks
Here, we have focused on the phenomenon by which proteins form open-ended assemblies in their folded state. A phenomenon that we termed agglomeration to differentiate it from other open-ended assembly phenomena with similar macroscopic features such as aggregation. We reviewed the main structural, chemical, and physical factors linked to protein agglomeration, as well as some of the implications of agglomeration in health, disease, and aging. We hope this review will contribute to raising awareness within the scientific community regarding the prevalence and significance of this phenomenon. We aim to inspire researchers to actively explore and characterize any potential instances of agglomeration they may come across.
References
Ahnert SE, Marsh JA, Hernández H, Robinson CV, Teichmann SA (2015) Principles of assembly reveal a periodic table of protein complexes. Science 350(6266):aaa2245. https://doi.org/10.1126/science.aaa2245
Alberti S, Halfmann R, King O, Kapila A, Lindquist S (2009) A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 137(1):146–158. https://doi.org/10.1016/j.cell.2009.02.044
Alberti S, Hyman AA (2021) Biomolecular condensates at the nexus of cellular stress, protein aggregation disease and ageing. Nat Rev Mol Cell Biol 22(3):196–213. https://doi.org/10.1038/s41580-020-00326-6
Alberts B, Johnson A, Lewis J, Walter P, Raff M, Roberts K (2002) Molecular Biology of the Cell 4th Edition: International Student Edition. Routledge https://play.google.com/store/books/details?id=ozigkQEACAAJ
Alex JM, Rennie ML, Engilberge S, Lehoczki G, Dorottya H, Fizil Á, Batta G, Crowley PB (2019) Calixarene-mediated assembly of a small antifungal protein. IUCrJ 6(Pt 2):238–247. https://doi.org/10.1107/S2052252519000411
André I, Strauss CEM, Kaplan DB, Bradley P, Baker D (2008) Emergence of symmetry in homooligomeric biological assemblies. Proc Natl Acad Sci USA 105(42):16148–16152. https://doi.org/10.1073/pnas.0807576105
Andréll J, Hicks MG, Palmer T, Carpenter EP, Iwata S, Maher MJ (2009) Crystal structure of the acid-induced arginine decarboxylase from Escherichia coli: reversible decamer assembly controls enzyme activity. Biochemistry 48(18):3915–3927. https://doi.org/10.1021/bi900075d
Anthony SA, Burrell AL, Johnson MC, Duong-Ly KC, Kuo Y-M, Simonet JC, Michener P, Andrews A, Kollman JM, Peterson JR (2017) Reconstituted IMPDH polymers accommodate both catalytically active and inactive conformations. Mol Biol Cell. https://doi.org/10.1091/mbc.E17-04-0263
Arakawa T, Timasheff SN (1984) Mechanism of protein salting in and salting out by divalent cation salts: balance between hydration and salt binding. Biochemistry 23(25):5912–5923. https://www.ncbi.nlm.nih.gov/pubmed/6525340. https://pubs.acs.org/doi/abs/10.1021/bi00320a004
Arakawa T, Timasheff SN (1985) Theory of protein solubility. Methods Enzymol 114:49–77. https://doi.org/10.1016/0076-6879(85)14005-x
Aumiller WM, Davis BW, Keating CD (2014) Chapter Five — Phase separation as a possible means of nuclear compartmentalization. In: Hancock R, Jeon KW (eds) International review of cell and molecular biology, vol 307. Academic Press, pp 109–149. https://doi.org/10.1016/B978-0-12-800046-5.00005-9
Banani SF, Lee HO, Hyman AA, Rosen MK (2017) Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol 18(5):285–298. https://doi.org/10.1038/nrm.2017.7
Barry RM, Bitbol A-F, Lorestani A, Charles EJ, Habrian CH, Hansen JM, Li H-J, Baldwin EP, Wingreen NS, Kollman JM, Gitai Z (2014) Large-scale filament formation inhibits the activity of CTP synthetase. eLife 3:e03638. https://doi.org/10.7554/eLife.03638
Beaty NB, Lane MD (1983) The polymerization of acetyl-CoA carboxylase. J Biol Chem 258(21):13051–13055 https://www.ncbi.nlm.nih.gov/pubmed/6138356
Behe MJ, Englander SW (1978) Sickle hemoglobin gelation. Reaction order and critical nucleus size. Biophys J 23(1):129–145. https://doi.org/10.1016/S0006-3495(78)85438-1
Boatz JC, Whitley MJ, Li M, Gronenborn AM, van der Wel PCA (2017) Cataract-associated P23T γD-crystallin retains a native-like fold in amorphous-looking aggregates formed at physiological pH. Nat Commun 8:15137. https://doi.org/10.1038/ncomms15137
Boye JI, Alli I, Ismail AA (1996) Interactions involved in the gelation of bovine serum Albumin. J Agric Food Chem 44(4):996–1004. https://doi.org/10.1021/jf950529t
Brangwynne CP, Eckmann CR, Courson DS, Rybarska A, Hoege C, Gharakhani J, Jülicher F, Hyman AA (2009) Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 324(5935):1729–1732. https://doi.org/10.1126/science.1172046
Brodin JD, Ambroggio XI, Tang C, Parent KN, Baker TS, Tezcan FA (2012) Metal-directed, chemically tunable assembly of one-, two- and three-dimensional crystalline protein arrays. Nat Chem 4(5):375–382. https://doi.org/10.1038/nchem.1290
Brubaker WD, Freites JA, Golchert KJ, Shapiro RA, Morikis V, Tobias DJ, Martin RW (2011) Separating instability from aggregation propensity in γS-crystallin variants. Biophys J 100(2):498–506. https://doi.org/10.1016/j.bpj.2010.12.3691
Buitink J, Leprince O (2004) Glass formation in plant anhydrobiotes: survival in the dry state. Cryobiology 48(3):215–228. https://doi.org/10.1016/j.cryobiol.2004.02.011
Bukau B, Weissman J, Horwich A (2006) Molecular chaperones and protein quality control. Cell 125(3):443–451. https://doi.org/10.1016/j.cell.2006.04.014
Burazerovic S, Gradinaru J, Pierron J, Ward TR (2007) Hierarchical self-assembly of one-dimensional streptavidin bundles as a collagen mimetic for the biomineralization of calcite. Angewandte Chemie 46(29):5510–5514. https://doi.org/10.1002/anie.200701080
Burrell AL, Nie C, Said M, Simonet JC, Fernández-Justel D, Johnson MC, Quispe J, Buey RM, Peterson JR, Kollman JM (2022) IMPDH1 retinal variants control filament architecture to tune allosteric regulation. Nat Struct Mol Biol 29(1):47–58. https://doi.org/10.1038/s41594-021-00706-2
Busch A, Waksman G (2012) Chaperone-usher pathways: diversity and pilus assembly mechanism. Philos Trans Royal Soc London. Series B, Biol Sci 367(1592):1112–1122. https://doi.org/10.1098/rstb.2011.0206
Chang C-C, Lin W-C, Pai L-M, Lee H-S, Wu S-C, Ding S-T, Liu J-L, Sung L-Y (2015) Cytoophidium assembly reflects upregulation of IMPDH activity. J Cell Sci 128(19):3550–3555. https://doi.org/10.1242/jcs.175265
Charache S, Conley CL, Waugh DF, Ugoretz RJ, Spurrell JR (1967) Pathogenesis of hemolytic anemia in homozygous hemoglobin C disease. J Clin Investig 46(11):1795–1811. https://doi.org/10.1172/JCI105670
Cherkasov V, Hofmann S, Druffel-Augustin S, Mogk A, Tyedmers J, Stoecklin G, Bukau B (2013) Coordination of translational control and protein homeostasis during severe heat stress. Curr Biol: CB 23(24):2452–2462. https://doi.org/10.1016/j.cub.2013.09.058
Chiti F, Dobson CM (2006) Protein misfolding, functional amyloid, and human disease. Ann Rev Biochem 75:333–366. https://doi.org/10.1146/annurev.biochem.75.101304.123901
Chiti F, Dobson CM (2017) Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Ann Rev Biochem 86:27–68. https://doi.org/10.1146/annurev-biochem-061516-045115
Chothia C, Janin J (1975) Principles of protein–protein recognition. Nature 256(5520):705–708. https://doi.org/10.1038/256705a0
Constantinescu P, Brown RA, Wyatt AR, Ranson M, Wilson MR (2017) Amorphous protein aggregates stimulate plasminogen activation, leading to release of cytotoxic fragments that are clients for extracellular chaperones. J Biol Chem 292(35):14425–14437. https://doi.org/10.1074/jbc.M117.786657
Curk T, Dobnikar J, Frenkel D (2017) Design principles for super selectivity using multivalent interactions. In: Multivalency. John Wiley & Sons, Ltd, pp 75–101. https://doi.org/10.1002/9781119143505.ch3
Decker CJ, Parker R (2012) P-bodies and stress granules: possible roles in the control of translation and mRNA degradation. Cold Spring Harbor Perspect Biol 4(9):a012286. https://doi.org/10.1101/cshperspect.a012286
Delarue M, Brittingham GP, Pfeffer S, Surovtsev IV, Pinglay S, Kennedy KJ, Schaffer M, Gutierrez JI, Sang D, Poterewicz G, Chung JK, Plitzko JM, Groves JT, Jacobs-Wagner C, Engel BD, Holt LJ (2018) mTORC1 controls phase separation and the biophysical properties of the cytoplasm by tuning crowding. Cell 174(2):338–349.e20. https://doi.org/10.1016/j.cell.2018.05.042
Derewenda ZS (2004) Rational protein crystallization by mutational surface engineering. Structure 12(4):529–535. https://doi.org/10.1016/j.str.2004.03.008
Dobson CM (2003) Protein folding and misfolding. Nature 426(6968):884–890. https://doi.org/10.1038/nature02261
Doye JPK, Louis AA, Vendruscolo M (2004) Inhibition of protein crystallization by evolutionary negative design. Phys Biol 1(1-2):P9–P13. https://doi.org/10.1088/1478-3967/1/1/P02
Drenckhahn D, Pollard TD (1986) Elongation of actin filaments is a diffusion-limited reaction at the barbed end and is accelerated by inert macromolecules. J Biol Chem 261(27):12754–12758. https://www.ncbi.nlm.nih.gov/pubmed/3745211. https://doi.org/10.1016/S0021-9258(18)67157-1
Duong-Ly KC, Gabelli SB (2014) Salting out of proteins using ammonium sulfate precipitation. Methods Enzymol 541:85–94. https://doi.org/10.1016/B978-0-12-420119-4.00007-0
Eaton WA, Hofrichter J (1990) Sickle cell hemoglobin polymerization. Adv Protein Chem 40:63–279. https://www.ncbi.nlm.nih.gov/pubmed/2195851. https://doi.org/10.1016/S0065-3233(08)60287-9
Empereur-Mot C, Garcia-Seisdedos H, Elad N, Dey S, Levy ED (2019) Geometric description of self-interaction potential in symmetric protein complexes. Sci Data 6(1):64. https://doi.org/10.1038/s41597-019-0058-x
Fahien LA, MacDonald MJ, Teller JK, Fibich B, Fahien CM (1989) Kinetic advantages of hetero-enzyme complexes with glutamate dehydrogenase and the α-ketoglutarate dehydrogenase complex. J Biol Chem 264(21):12303–12312. https://doi.org/10.1016/S0021-9258(18)63859-1
Fasting C, Schalley CA, Weber M, Seitz O, Hecht S, Koksch B, Dernedde J, Graf C, Knapp E-W, Haag R (2012) Multivalency as a chemical organization and action principle. Angewandte Chemie 51(42):10472–10498. https://doi.org/10.1002/anie.201201114
Ferreira APS, Cassago A, de Almeida Gonçalves K, Dias MM, Adamoski D, Ascenção CFR, Honorato RV, de Oliveira JF, Ferreira IM, Fornezari C, Bettini J (2013) Active glutaminase C self-assembles into a supratetrameric oligomer that can be disrupted by an allosteric inhibitor. J Biol Chem 288(39):28009–28020. https://doi.org/10.1074/jbc.M113.501346
Fink AL (1998) Protein aggregation: folding aggregates, inclusion bodies and amyloid. Fold Design 3(1):R9–R23. https://doi.org/10.1016/S1359-0278(98)00002-9
Flory PJ (1953) Principles of polymer chemistry. Cornell University Press. https://play.google.com/store/books/details?id=CQ0EbEkT5R0C. https://doi.org/10.1126/science.119.3095.555.b
Franzmann TM, Alberti S (2019) Prion-like low-complexity sequences: key regulators of protein solubility and phase behavior. J Biol Chem 294(18):7128–7136. https://doi.org/10.1074/jbc.TM118.001190
Franzmann TM, Jahnel M, Pozniakovsky A, Mahamid J, Holehouse AS, Nüske E, Richter D, Baumeister W, Grill SW, Pappu RV, Hyman AA, Alberti S (2018) Phase separation of a yeast prion protein promotes cellular fitness. Science 359(6371). https://doi.org/10.1126/science.aao5654
Frieden C (1959) Glutamic dehydrogenase. II. The effect of various nucleotides on the association-dissociation and kinetic properties. J Biol Chem 234(4):815–820 https://www.ncbi.nlm.nih.gov/pubmed/13654269
Garcia Seisdedos H, Levin T, Shapira G, Freud S, Levy ED (2022) Mutant libraries reveal negative design shielding proteins from supramolecular self-assembly and relocalization in cells. Proc Natl Acad Sci USA 119(5). https://doi.org/10.1073/pnas.2101117119
Garcia-Seisdedos H, Empereur-Mot C, Elad N, Levy ED (2017) Proteins evolve on the edge of supramolecular self-assembly. Nature 548(7666):244–247. https://doi.org/10.1038/nature23320
Garcia-Seisdedos H, Heidenreich M, Levy ED (2020) Not going with the flow: how cells adapt internal physics [Review of Not Going with the Flow: How Cells Adapt Internal Physics]. Cell 183(6):1462–1463. https://doi.org/10.1016/j.cell.2020.11.021
Garcia-Seisdedos H, Ibarra-Molero B, Sanchez-Ruiz JM (2012) How many ionizable groups can sit on a protein hydrophobic core? Proteins 80(1):1–7. https://doi.org/10.1002/prot.23166
Garcia-Seisdedos, H., Villegas, J. A., & Levy, E. D. (2019) Infinite assembly of folded proteins in evolution, disease, and engineering. In Angewandte Chemie International Edition (58, 17, pp. 5514–5531). https://doi.org/10.1002/anie.201806092
Geiler-Samerotte KA, Dion MF, Budnik BA, Wang SM, Hartl DL, Drummond DA (2011) Misfolded proteins impose a dosage-dependent fitness cost and trigger a cytosolic unfolded protein response in yeast. Proc Natl Acad Sci USA 108(2):680–685. https://doi.org/10.1073/pnas.1017570108
Gibb BC (2019) Hofmeister’s curse. Nat Chem 11(11):963–965. https://doi.org/10.1038/s41557-019-0355-1
Gomes E, Shorter J (2019) The molecular language of membraneless organelles. J Biol Chem 294(18):7115–7127. https://doi.org/10.1074/jbc.TM118.001192
Gonen S, DiMaio F, Gonen T, Baker D (2015) Design of ordered two-dimensional arrays mediated by noncovalent protein-protein interfaces. Science 348(6241):1365–1368. https://doi.org/10.1126/science.aaa9897
Goodsell DS, Olson AJ (2000) Structural symmetry and protein function. Ann Rev Biophys Biomol Struct 29:105–153. https://doi.org/10.1146/annurev.biophys.29.1.105
Gremer L, Schölzel D, Schenk C, Reinartz E, Labahn J, Ravelli RBG, Tusche M, Lopez-Iglesias C, Hoyer W, Heise H, Willbold D, Schröder GF (2017) Fibril structure of amyloid-β(1–42) by cryo–electron microscopy. Science 358(6359):116–119. https://doi.org/10.1126/science.aao2825
Grueninger D, Treiber N, Ziegler MOP, Koetter JWA, Schulze M-S, Schulz GE (2008) Designed protein-protein association. Science 319(5860):206–209. https://doi.org/10.1126/science.1150421
Guo Z, Eisenberg D (2006) Runaway domain swapping in amyloid-like fibrils of T7 endonuclease I. Proc Natl Acad Sci USA 103(21):8042–8047. https://doi.org/10.1073/pnas.0602607103
Guyer MF, Claus PE (1942) Increased viscosity of cells of induced tumors*. Cancer Res 2(1):16–18. https://aacrjournals.org/cancerres/article-abstract/2/1/16/474094
Gylfe E (1976) Comparison of the effects of leucines, non-metabolizable leucine analogues and other insulin secretagogues on the activity of glutamate dehydrogenase. Acta Diabetol Latina 13(1-2):20–24. https://doi.org/10.1007/BF02591577
Harrington DJ, Adachi K, Royer WE Jr (1997) The high resolution crystal structure of deoxyhemoglobin S. J Mol Biol 272(3):398–407. https://doi.org/10.1006/jmbi.1997.1253
Hatefi Y, Hanstein WG (1969) Solubilization of particulate proteins and nonelectrolytes by chaotropic agents. Proc Natl Acad Sci USA 62(4):1129–1136. https://doi.org/10.1073/pnas.62.4.1129
Heidenreich M, Georgeson JM, Locatelli E, Rovigatti L, Nandi SK, Steinberg A, Nadav Y, Shimoni E, Safran SA, Doye JPK, Levy ED (2020) Designer protein assemblies with tunable phase diagrams in living cells. Nat Chem Biol 16(9):939–945. https://doi.org/10.1038/s41589-020-0576-z
Herzog W, Weber K (1978) Microtubule formation by pure brain tubulin in vitro. The influence of dextran and poly(ethylene glycol). Eur J Biochem/FEBS 91(1):249–254. https://doi.org/10.1111/j.1432-1033.1978.tb20959.x
Hoeg-Jensen T, Havelund S, Nielsen PK, Markussen J (2005) Reversible insulin self-assembly under carbohydrate control. J Am Chem Soc 127(17):6158–6159. https://doi.org/10.1021/ja051038k
Hofmeister F (1888) Arbeiten aus dem pharmakologisehen Institut der deutschen Universität zu Prag. 12. Zur Lehre von der Wirkung der Salze. Archiv Für Experimentelle Pathologie Und Pharmakologie 25(1):1–30. https://doi.org/10.1007/BF01838161
Hunkeler M, Hagmann A, Stuttfeld E, Chami M, Guri Y, Stahlberg H, Maier T (2018) Structural basis for regulation of human acetyl-CoA carboxylase. Nature 558(7710):470–474. https://doi.org/10.1038/s41586-018-0201-4
Hvorecny KL, Kollman JM (2023) Greater than the sum of parts: mechanisms of metabolic regulation by enzyme filaments. Curr Opin Struct Biol 79:102530. https://doi.org/10.1016/j.sbi.2023.102530
Hyman AA, Weber CA, Jülicher F (2014) Liquid-liquid phase separation in biology. Ann Rev Cell Dev Biol 30:39–58. https://doi.org/10.1146/annurev-cellbio-100913-013325
Iadanza MG, Jackson MP, Hewitt EW, Ranson NA, Radford SE (2018) A new era for understanding amyloid structures and disease. Nat Rev Mol Cell Biol 19(12):755–773. https://doi.org/10.1038/s41580-018-0060-8
Ispolatov I, Yuryev A, Mazo I, Maslov S (2005) Binding properties and evolution of homodimers in protein-protein interaction networks. Nucleic Acids Res 33(11):3629–3635. https://doi.org/10.1093/nar/gki678
Joyner RP, Tang JH, Helenius J, Dultz E, Brune C, Holt LJ, Huet S, Müller DJ, Weis K (2016) A glucose-starvation response regulates the diffusion of macromolecules. eLife 5. https://doi.org/10.7554/eLife.09376
Kim C-W, Moon Y-A, Park SW, Cheng D, Kwon HJ, Horton JD (2010) Induced polymerization of mammalian acetyl-CoA carboxylase by MIG12 provides a tertiary level of regulation of fatty acid synthesis. Proc Natl Acad Sci USA 107(21):9626–9631. https://doi.org/10.1073/pnas.1001292107
Kim S-Y, Kim Y-W, Hegerl R, Cyrklaff M, Kim I-S (2005) Novel type of enzyme multimerization enhances substrate affinity of oat beta-glucosidase. J Struct Biol 150(1):1–10. https://doi.org/10.1016/j.jsb.2004.07.007
Kitagishi H, Oohora K, Yamaguchi H, Sato H, Matsuo T, Harada A, Hayashi T (2007) Supramolecular hemoprotein linear assembly by successive interprotein heme-heme pocket interactions. J Am Chem Soc 129(34):10326–10327. https://doi.org/10.1021/ja073295q
Kmoch S, Brynda J, Asfaw B, Bezouška K, Novák P, Řezáčová P, Ondrová L, Filipec M, Sedláček J, Elleder M (2000) Link between a novel human γD-crystallin allele and a unique cataract phenotype explained by protein crystallography. Hum Mol Genet 9(12):1779–1786. https://doi.org/10.1093/hmg/9.12.1779
Kramer RM, Shende VR, Motl N, Pace CN, Scholtz JM (2012) Toward a molecular understanding of protein solubility: increased negative surface charge correlates with increased solubility. Biophys J 102(8):1907–1915. https://doi.org/10.1016/j.bpj.2012.01.060
Krueger S, Nossal R (1988) SANS studies of interacting hemoglobin in intact erythrocytes. Biophys J 53(1):97–105. https://doi.org/10.1016/S0006-3495(88)83070-4
Labesse G, Alexandre T, Vaupré L, Salard-Arnaud I, Him JLK, Raynal B, Bron P, Munier-Lehmann H (2013) MgATP regulates allostery and fiber formation in IMPDHs. Structure 21(6):975–985. https://doi.org/10.1016/j.str.2013.03.011
Lanci CJ, MacDermaid CM, Kang S-G, Acharya R, North B, Yang X, Qiu XJ, DeGrado WF, Saven JG (2012) Computational design of a protein crystal. Proc Natl Acad Sci USA 109(19):7304–7309. https://doi.org/10.1073/pnas.1112595109
Lawson DM, Artymiuk PJ, Yewdall SJ, Smith JM, Livingstone JC, Treffry A, Luzzago A, Levi S, Arosio P, Cesareni G (1991) Solving the structure of human H ferritin by genetically engineering intermolecular crystal contacts. Nature 349(6309):541–544. https://doi.org/10.1038/349541a0
Levy ED (2010) A simple definition of structural regions in proteins and its use in analyzing interface evolution. J Mol Biol 403(4):660–670. https://doi.org/10.1016/j.jmb.2010.09.028
Levy ED, Boeri Erba E, Robinson CV, Teichmann SA (2008) Assembly reflects evolution of protein complexes. Nature 453(7199):1262–1265. https://doi.org/10.1038/nature06942
Levy, E. D., De, S., & Teichmann, S. A. (2012) Cellular crowding imposes global constraints on the chemistry and evolution of proteomes. In Proceedings of the National Academy of Sciences (109, 50, pp. 20461–20466). https://doi.org/10.1073/pnas.1209312109
Levy ED, Teichmann S (2013) Structural, evolutionary, and assembly principles of protein oligomerization. Prog Mol Biol Trans Sci 117:25–51. https://doi.org/10.1016/B978-0-12-386931-9.00002-7
Lindner RA, Ralston GB (1997) Macromolecular crowding: effects on actin polymerisation. Biophys Chem 66(1):57–66. https://doi.org/10.1016/s0301-4622(97)00011-2
Liu J-L (2016) The cytoophidium and its kind: filamentation and compartmentation of metabolic enzymes. Ann Rev Cell Dev Biol 32:349–372. https://doi.org/10.1146/annurev-cellbio-111315-124907
López-Alonso JP, Bruix M, Font J, Ribó M, Vilanova M, Jiménez MA, Santoro J, González C, Laurents DV (2010) NMR spectroscopy reveals that RNase A is chiefly denatured in 40% acetic acid: implications for oligomer formation by 3D domain swapping. J Am Chem Soc 132(5):1621–1630. https://doi.org/10.1021/ja9081638
Lukatsky DB, Shakhnovich BE, Mintseris J, Shakhnovich EI (2007) Structural similarity enhances interaction propensity of proteins. J Mol Biol 365(5):1596–1606. https://doi.org/10.1016/j.jmb.2006.11.020
Lukatsky DB, Zeldovich KB, Shakhnovich EI (2006) Statistically enhanced self-attraction of random patterns. Phys Rev Lett 97(17):178101. https://doi.org/10.1103/PhysRevLett.97.178101
Lynch EM, Hicks DR, Shepherd M, Endrizzi JA, Maker A, Hansen JM, Barry RM, Gitai Z, Baldwin EP, Kollman JM (2017) Human CTP synthase filament structure reveals the active enzyme conformation. Nat Struct Mol Biol 24(6):507–514. https://doi.org/10.1038/nsmb.3407
Lynch EM, Kollman JM (2020) Coupled structural transitions enable highly cooperative regulation of human CTPS2 filaments. Nat Struct Mol Biol 27(1):42–48. https://doi.org/10.1038/s41594-019-0352-5
Lynch EM, Kollman JM, Webb BA (2020) Filament formation by metabolic enzymes—a new twist on regulation. Curr Opin Cell Biol 66:28–33. https://doi.org/10.1016/j.ceb.2020.04.006
Marcus Y (2009) Effect of ions on the structure of water: structure making and breaking. Chem Rev 109(3):1346–1370. https://doi.org/10.1021/cr8003828
Marini G, Nüske E, Leng W, Alberti S, Pigino G (2020) Reorganization of budding yeast cytoplasm upon energy depletion. Mol Biol Cell 31(12):1232–1245. https://doi.org/10.1091/mbc.E20-02-0125
Matsudomi N, Rector D, Kinsella JE (1991) Gelation of bovine serum albumin and β-lactoglobulin; effects of pH, salts and thiol reagents. Food Chem 40(1):55–69. https://doi.org/10.1016/0308-8146(91)90019-K
Miermont A, Waharte F, Hu S, McClean MN, Bottani S, Léon S, Hersen P (2013) Severe osmotic compression triggers a slowdown of intracellular signaling, which can be explained by molecular crowding. Proc Natl Acad Sci USA 110(14):5725–5730. https://doi.org/10.1073/pnas.1215367110
Minsky A, Shimoni E, Frenkiel-Krispin D (2002) Stress, order and survival. Nat Rev Mol Cell Biol 3(1):50–60. https://doi.org/10.1038/nrm700
Minton AP (2000) Implications of macromolecular crowding for protein assembly. Curr Opin Struct Biol 10(1):34–39. https://doi.org/10.1016/s0959-440x(99)00045-7
Monod J, Wyman J, Changeux JP (1965) On the nature of allosteric transitions: a plausible model. J Mol Biol 12:88–118. https://doi.org/10.1016/s0022-2836(65)80285-6
Montrose K, López Cabezas RM, Paukštytė J, Saarikangas J (2020) Winter is coming: regulation of cellular metabolism by enzyme polymerization in dormancy and disease. Exp Cell Res 397(2):112383. https://doi.org/10.1016/j.yexcr.2020.112383
Moon JC, Hah YS, Kim WY, Jung BG, Jang HH (2005a) Oxidative stress-dependent structural and functional switching of a human 2-Cys peroxiredoxin isotype II that enhances HeLa cell resistance to H2O2-induced …. J Biol https://www.jbc.org/article/S0021-9258(20)56579-4/abstract. https://doi.org/10.1074/jbc.M505362200
Moon JC, Hah Y-S, Kim WY, Jung BG, Jang HH, Lee JR, Kim SY, Lee YM, Jeon MG, Kim CW, Others. (2005b) Oxidative stress-dependent structural and functional switching of a human 2-Cys peroxiredoxin isotype II that enhances HeLa cell resistance to H2O2-induced cell death. J Biol Chem 280(31):28775–28784. https://www.jbc.org/article/S0021-9258(20)56579-4/abstract. https://doi.org/10.1074/jbc.M505362200
Mou Y, Yu J-Y, Wannier TM, Guo C-L, Mayo SL (2015) Computational design of co-assembling protein–DNA nanowires. Nature 525(7568):230–233. https://doi.org/10.1038/nature14874
Müller MK, Petkau K, Brunsveld L (2011) Protein assembly along a supramolecular wire. Chem Commun 47(1):310–312. https://doi.org/10.1039/c0cc02084b
Munder MC, Midtvedt D, Franzmann T, Nüske E, Otto O, Herbig M, Ulbricht E, Müller P, Taubenberger A, Maharana S, Malinovska L, Richter D, Guck J, Zaburdaev V, Alberti S (2016) A pH-driven transition of the cytoplasm from a fluid- to a solid-like state promotes entry into dormancy. eLife 5. https://doi.org/10.7554/elife.09347
Narayanaswamy R, Levy M, Tsechansky M, Stovall GM, O’Connell JD, Mirrielees J, Ellington AD, Marcotte EM (2009) Widespread reorganization of metabolic enzymes into reversible assemblies upon nutrient starvation. Proc Natl Acad Sci USA 106(25):10147–10152. https://doi.org/10.1073/pnas.0812771106
Nüske E, Marini G, Richter D, Leng W, Bogdanova A, Franzmann TM, Pigino G, Alberti S (2020) Filament formation by the translation factor eIF2B regulates protein synthesis in starved cells. Biol Open 9(7). https://doi.org/10.1242/bio.046391
O’Brien L, Shelley K, Towfighi J, McPherson A (1980) Crystalline ribosomes are present in brains from senile humans. Proc Natl Acad Sci USA 77(4):2260–2264. https://doi.org/10.1073/pnas.77.4.2260
Orij R, Urbanus ML, Vizeacoumar FJ, Giaever G, Boone C, Nislow C, Brul S, Smits GJ (2012) Genome-wide analysis of intracellular pH reveals quantitative control of cell division rate by pHc in Saccharomyces cerevisiae. Genome Biol 13(9):R80. https://doi.org/10.1186/gb-2012-13-9-r80
Padilla JE, Colovos C, Yeates TO (2001) Nanohedra: using symmetry to design self assembling protein cages, layers, crystals, and filaments. Proc Natl Acad Sci USA 98(5):2217–2221. https://doi.org/10.1073/pnas.041614998
Pak CW, Kosno M, Holehouse AS, Padrick SB, Mittal A, Ali R, Yunus AA, Liu DR, Pappu RV, Rosen MK (2016) Sequence determinants of intracellular phase separation by complex coacervation of a disordered protein. Mol Cell 63(1):72–85. https://doi.org/10.1016/j.molcel.2016.05.042
Pande A, Annunziata O, Asherie N, Ogun O, Benedek GB, Pande J (2005) Decrease in protein solubility and cataract formation caused by the Pro23 to Thr mutation in human gamma D-crystallin. Biochemistry 44(7):2491–2500. https://doi.org/10.1021/bi0479611
Park CK, Horton NC (2019) Structures, functions, and mechanisms of filament forming enzymes: a renaissance of enzyme filamentation. Biophys Rev 11(6):927–994. https://doi.org/10.1007/s12551-019-00602-6
Park YC, Burkitt V, Villa AR, Tong L, Wu H (1999) Structural basis for self-association and receptor recognition of human TRAF2. Nature 398(6727):533–538. https://doi.org/10.1038/19110
Parker R, Noel TR, Brownsey GJ, Laos K, Ring SG (2005) The nonequilibrium phase and glass transition behavior of beta-lactoglobulin. Biophys J 89(2):1227–1236. https://doi.org/10.1529/biophysj.105.064246
Parry BR, Surovtsev IV, Cabeen MT, O’Hern CS, Dufresne ER, Jacobs-Wagner C (2014) The bacterial cytoplasm has glass-like properties and is fluidized by metabolic activity. Cell 156(1-2):183–194. https://doi.org/10.1016/j.cell.2013.11.028
Patel A, Malinovska L, Saha S, Wang J, Alberti S, Krishnan Y, Hyman AA (2017) ATP as a biological hydrotrope. Science 356(6339):753–756. https://doi.org/10.1126/science.aaf6846
Paukštytė J, López Cabezas RM, Feng Y, Tong K, Schnyder D, Elomaa E, Gregorova P, Doudin M, Särkkä M, Sarameri J, Lippi A, Vihinen H, Juutila J, Nieminen A, Törönen P, Holm L, Jokitalo E, Krisko A, Huiskonen J et al (2023) Global analysis of aging-related protein structural changes uncovers enzyme-polymerization-based control of longevity. Mol Cell. https://doi.org/10.1016/j.molcel.2023.08.015
Pechmann S, Levy ED, Tartaglia GG, Vendruscolo M (2009) Physicochemical principles that regulate the competition between functional and dysfunctional association of proteins. Proc Natl Acad Sci USA 106(25):10159–10164. https://doi.org/10.1073/pnas.0812414106
Persson LB, Ambati VS, Brandman O (2020) Cellular control of viscosity counters changes in temperature and energy availability. Cell 183(6):1572–1585.e16. https://doi.org/10.1016/j.cell.2020.10.017
Petkau-Milroy K, Sonntag MH, Colditz A, Brunsveld L (2013) Multivalent protein assembly using monovalent self-assembling building blocks. Int J Mol Sci 14(10):21189–21201. https://doi.org/10.3390/ijms141021189
Petrovska I, Nüske E, Munder MC, Kulasegaran G, Malinovska L, Kroschwald S, Richter D, Fahmy K, Gibson K, Verbavatz J-M, Alberti S (2014) Filament formation by metabolic enzymes is a specific adaptation to an advanced state of cellular starvation. eLife. https://doi.org/10.7554/eLife.02409
Pintsch T, Satre M, Klein G, Martin JB, Schuster SC (2001) Cytosolic acidification as a signal mediating hyperosmotic stress responses in Dictyostelium discoideum. BMC Cell Biol 2:9. https://doi.org/10.1186/1471-2121-2-9
Pratt AJ, Shin DS, Merz GE, Rambo RP, Lancaster WA, Dyer KN, Borbat PP, Poole FL 2nd, Adams MWW, Freed JH, Crane BR, Tainer JA, Getzoff ED (2014) Aggregation propensities of superoxide dismutase G93 hotspot mutants mirror ALS clinical phenotypes. Proc Natl Acad Sci USA 111(43):E4568–E4576. https://doi.org/10.1073/pnas.1308531111
Prouteau M, Desfosses A, Sieben C, Bourgoint C, Lydia Mozaffari N, Demurtas D, Mitra AK, Guichard P, Manley S, Loewith R (2017) TORC1 organized in inhibited domains (TOROIDs) regulate TORC1 activity. Nature 550(7675):265–269. https://doi.org/10.1038/nature24021
Ralston GB (1990) Effects of “crowding” in protein solutions. J Chem Educ 67(10):857. https://doi.org/10.1021/ed067p857
Renard D, Lefebvre J (1992) Gelation of globular proteins: effect of pH and ionic strength on the critical concentration for gel formation. A simple model and its application to beta-lactoglobulin heat-induced gelation. Int J Biol Macromol 14(5):287–291. https://doi.org/10.1016/s0141-8130(05)80042-x
Riback JA, Katanski CD, Kear-Scott JL, Pilipenko EV, Rojek AE, Sosnick TR, Drummond DA (2017) Stress-triggered phase separation is an adaptive, evolutionarily tuned response. Cell 168(6):1028–1040.e19. https://doi.org/10.1016/j.cell.2017.02.027
Romero ML, Garcia Seisdedos H, Ibarra-Molero B (2022) Active site center redesign increases protein stability preserving catalysis in thioredoxin. Protein Sci: Publication Protein Soc 31(9). https://doi.org/10.1002/pro.4417
Romero Romero ML, Yang F, Lin Y-R, Toth-Petroczy A, Berezovsky IN, Goncearenco A, Yang W, Wellner A, Kumar-Deshmukh F, Sharon M, Baker D, Varani G, Tawfik DS (2018) Simple yet functional phosphate-loop proteins. Proc Natl Acad Sci USA 115(51):E11943–E11950. https://doi.org/10.1073/pnas.1812400115
Rosenzweig R, Nillegoda NB, Mayer MP, Bukau B (2019) The Hsp70 chaperone network. Nat Rev Mol Cell Biol 20(11):665–680. https://doi.org/10.1038/s41580-019-0133-3
Ross PD, Minton AP (1979) The effect of non-aggregating proteins upon the gelation of sickle cell hemoglobin: model calculations and data analysis. Biochem Biophys Res Commun 88(4):1308–1314. https://doi.org/10.1016/0006-291x(79)91123-9
Rousset M, Zweibaum A, Fogh J (1981) Presence of glycogen and growth-related variations in 58 cultured human tumor cell lines of various tissue origins. Cancer Res 41(3):1165–1170. https://www.ncbi.nlm.nih.gov/pubmed/7459858
Saccoccia F, Di Micco P, Boumis G, Brunori M, Koutris I, Miele AE, Morea V, Sriratana P, Williams DL, Bellelli A, Angelucci F (2012) Moonlighting by different stressors: crystal structure of the chaperone species of a 2-Cys peroxiredoxin. Structure 20(3):429–439. https://doi.org/10.1016/j.str.2012.01.004
Scatchard, G., Cohn, E. J., Edsall, J. T., Cohn, E. J., Hans Mueller (de l’Institut de technologie du Massachussett), Oncley, J. L., & Kirkwood, G. (1943) Proteins, amino acids and peptides as ions and dipolar ions: Edwin J. Cohn and John T. Edsall ... Including, G. Kirkwood ... Hans Mueller ... J.L. Oncley ... George Scatchard. Reinhold (Waverly Press). https://play.google.com/store/books/details?id=1qMazQEACAAJ
Schulz GE (2010) The dominance of symmetry in the evolution of homo-oligomeric proteins. J Mol Biol 395(4):834–843. https://doi.org/10.1016/j.jmb.2009.10.044
Schwartz R, Ting CS, King J (2001) Whole proteome pI values correlate with subcellular localizations of proteins for organisms within the three domains of life. Genome Res 11(5):703–709. https://doi.org/10.1101/gr.gr-1587r
Sigler PB, Xu Z, Rye HS, Burston SG, Fenton WA, Horwich AL (1998) Structure and function in GroEL-mediated protein folding. Ann Rev Biochem 67:581–608. https://doi.org/10.1146/annurev.biochem.67.1.581
Simonet JC, Burrell AL, Kollman JM, Peterson JR (2020) Freedom of assembly: metabolic enzymes come together. Mol Biol Cell 31(12):1201–1205. https://doi.org/10.1091/mbc.E18-10-0675
Słabicki M, Yoon H, Koeppel J, Nitsch L, Roy Burman SS, Di Genua C, Donovan KA, Sperling AS, Hunkeler M, Tsai JM, Sharma R, Guirguis A, Zou C, Chudasama P, Gasser JA, Miller PG, Scholl C, Fröhling S, Nowak RP et al (2020) Small-molecule-induced polymerization triggers degradation of BCL6. Nature 588(7836):164–168. https://doi.org/10.1038/s41586-020-2925-1
Song WJ, Sontz PA, Ambroggio XI, Tezcan FA (2014) Metals in protein–protein interfaces. Ann Rev Biophys 43(1):409–431. https://doi.org/10.1146/annurev-biophys-051013-023038
Song WJ, Tezcan FA (2014) A designed supramolecular protein assembly with in vivo enzymatic activity. Science 346(6216):1525–1528. https://doi.org/10.1126/science.1259680
Sontag EM, Vonk WIM, Frydman J (2014) Sorting out the trash: the spatial nature of eukaryotic protein quality control. Curr Opin Cell Biol 26:139–146. https://doi.org/10.1016/j.ceb.2013.12.006
Stoddard PR, Lynch EM, Farrell DP, Dosey AM, DiMaio F, Williams TA, Kollman JM, Murray AW, Garner EC (2020) Polymerization in the actin ATPase clan regulates hexokinase activity in yeast. Science 367(6481):1039–1042. https://doi.org/10.1126/science.aay5359
Sukegawa H, Nishimura T, Yoshio M, Kajiyama S, Kato T (2017) One-dimensional supramolecular hybrids: self-assembled nanofibrous materials based on a sugar gelator and calcite developed along an unusual axis. CrystEngComm/RSC 19(12):1580–1584. https://doi.org/10.1039/C7CE00140A
Suzuki Y, Cardone G, Restrepo D, Zavattieri PD, Baker TS, Tezcan FA (2016) Self-assembly of coherently dynamic, auxetic, two-dimensional protein crystals. Nature 533(7603):369–373. https://doi.org/10.1038/nature17633
Takahashi S, Satomi A, Yano K, Kawase H, Tanimizu T, Tuji Y, Murakami S, Hirayama R (1999) Estimation of glycogen levels in human colorectal cancer tissue: relationship with cell cycle and tumor outgrowth. J Gastroenterol 34(4):474–480. https://doi.org/10.1007/s005350050299
Tanford C (1961) Physical Chemistry of Macromolecules. Wiley. https://play.google.com/store/books/details?id=gjlRAAAAMAAJ. https://doi.org/10.1002/pol.1962.1206217338
Tartaglia GG, Pechmann S, Dobson CM, Vendruscolo M (2007) Life on the edge: a link between gene expression levels and aggregation rates of human proteins. Trends Biochem Sci 32(5):204–206. https://doi.org/10.1016/j.tibs.2007.03.005
Tavenor NA, Murnin MJ, Horne WS (2017) Supramolecular metal-coordination polymers, nets, and frameworks from synthetic coiled-coil peptides. J Am Chem Soc 139(6):2212–2215. https://doi.org/10.1021/jacs.7b00651
Tellam RL, Sculley MJ, Nichol LW, Wills PR (1983) The influence of poly(ethylene glycol) 6000 on the properties of skeletal-muscle actin. Biochem J 213(3):651–659. https://doi.org/10.1042/bj2130651
Thomas EC, Gunter JH, Webster JA, Schieber NL, Oorschot V, Parton RG, Whitehead JP (2012) Different characteristics and nucleotide binding properties of inosine monophosphate dehydrogenase (IMPDH) isoforms. PloS One 7(12):e51096. https://doi.org/10.1371/journal.pone.0051096
Trappe V, Prasad V, Cipelletti L, Segre PN, Weitz DA (2001) Jamming phase diagram for attractive particles. Nature 411(6839):772–775. https://doi.org/10.1038/35081021
Valley CC, Cembran A, Perlmutter JD, Lewis AK, Labello NP, Gao J, Sachs JN (2012) The methionine-aromatic motif plays a unique role in stabilizing protein structure. J Biol Chem 287(42):34979–34991. https://doi.org/10.1074/jbc.M112.374504
Vecchi G, Sormanni P, Mannini B, Vandelli A, Tartaglia GG, Dobson CM, Hartl FU, Vendruscolo M (2020) Proteome-wide observation of the phenomenon of life on the edge of solubility. Proc Natl Acad Sci USA 117(2):1015–1020. https://doi.org/10.1073/pnas.1910444117
Vekilov PG, Feeling-Taylor AR, Petsev DN, Galkin O, Nagel RL, Hirsch RE (2002) Intermolecular interactions, nucleation, and thermodynamics of crystallization of hemoglobin C. Biophys J 83(2):1147–1156. https://doi.org/10.1016/S0006-3495(02)75238-7
Villegas JA, Heidenreich M, Levy ED (2022) Molecular and environmental determinants of biomolecular condensate formation. Nat Chem Biol 18(12):1319–1329. https://doi.org/10.1038/s41589-022-01175-4
Wales DJ (1998) Symmetry, near-symmetry and energetics. Chem Phys Lett 285(5):330–336. https://doi.org/10.1016/S0009-2614(98)00044-X
Wallace EWJ, Kear-Scott JL, Pilipenko EV, Schwartz MH, Laskowski PR, Rojek AE, Katanski CD, Riback JA, Dion MF, Franks AM, Airoldi EM, Pan T, Budnik BA, Drummond DA (2015) Reversible, specific, active aggregates of endogenous proteins assemble upon heat stress. Cell 162(6):1286–1298. https://doi.org/10.1016/j.cell.2015.08.041
Weitzel G, Pilatus U, Rensing L (1985) Similar dose response of heat shock protein synthesis and intracellular pH change in yeast. Exp Cell Res 159(1):252–256. https://doi.org/10.1016/s0014-4827(85)80054-9
Wilf J, Gladner JA, Minton AP (1985) Acceleration of fibrin gel formation by unrelated proteins. Thrombosis Res 37(6):681–688. https://doi.org/10.1016/0049-3848(85)90197-5
Wingfield P (2001) Protein precipitation using ammonium sulfate. Current Protocols in Protein Science / Editorial Board, John E. Coligan ... [et Al.], Appendix 3, Appendix 3F. https://doi.org/10.1002/0471140864.psa03fs13
Woodruff JB, Ferreira Gomes B, Widlund PO, Mahamid J, Honigmann A, Hyman AA (2017) The centrosome is a selective condensate that nucleates microtubules by concentrating tubulin. Cell 169(6):1066–1077.e10. https://doi.org/10.1016/j.cell.2017.05.028
Wright CF, Teichmann SA, Clarke J, Dobson CM (2005) The importance of sequence diversity in the aggregation and evolution of proteins. Nature 438(7069):878–881. https://doi.org/10.1038/nature04195
Wright PE, Dyson HJ (2015) Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol 16(1):18–29. https://doi.org/10.1038/nrm3920
Wu Y, Shu W, Zeng C, Guo B, Shi J, Jing J, Zhang X (2019) A mitochondria targetable and viscosity sensitive fluorescent probe and its applications for distinguishing cancerous cells. Dyes Pigments 168:134–139. https://doi.org/10.1016/j.dyepig.2019.04.049
Yang J-R, Liao B-Y, Zhuang S-M, Zhang J (2012) Protein misinteraction avoidance causes highly expressed proteins to evolve slowly. Proc Natl Acad Sci USA 109(14):E831–E840. https://doi.org/10.1073/pnas.1117408109
Yang M, Song WJ (2019) Diverse protein assembly driven by metal and chelating amino acids with selectivity and tunability. Nat Commun 10(1):5545. https://doi.org/10.1038/s41467-019-13491-w
Yeates TO (2017) Geometric principles for designing highly symmetric self-assembling protein nanomaterials. Ann Rev Biophys 46:23–42. https://doi.org/10.1146/annurev-biophys-070816-033928
Zhao H, Olubajo O, Song Z, Sims AL, Person TE, Lawal RA, Holley LA (2006) Effect of kosmotropicity of ionic liquids on the enzyme stability in aqueous solutions. Bioorg Chem 34(1):15–25. https://doi.org/10.1016/j.bioorg.2005.10.004
Zhou H-X, Rivas G, Minton AP (2008) Macromolecular crowding and confinement: biochemical, biophysical, and potential physiological consequences. Ann Rev Biophys 37:375–397. https://doi.org/10.1146/annurev.biophys.37.032807.125817
Zimmerman SB, Minton AP (1993) Macromolecular crowding: biochemical, biophysical, and physiological consequences. Ann Rev Biophys Biomol Struct 22:27–65. https://doi.org/10.1146/annurev.bb.22.060193.000331
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature. This work is supported by a research grant from the Spanish Ministry of Science and Innovation (Grant number PID2021-127150NA-I00). HGS is supported by a Ramon y Cajal fellowship from the Spanish Ministry of Science and Innovation (Grant number: RYC2020-030700-I).
Author information
Authors and Affiliations
Contributions
HGS conceived the research. MLRR and HGS performed the bibliographical research. MLRR and HGS wrote the manuscript.
Corresponding author
Ethics declarations
Ethics approval
This is an observational study. The CSIC Research Ethics Committee has confirmed that no ethical approval is required
Consent to participate and consent to publish
All images used in this manuscript have been reused from prior publications which obtained all the relevant information regarding consent.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Romero-Romero, M.L., Garcia-Seisdedos, H. Agglomeration: when folded proteins clump together. Biophys Rev 15, 1987–2003 (2023). https://doi.org/10.1007/s12551-023-01172-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12551-023-01172-4