
Arsinothricin Inhibits Plasmodium falciparum Proliferation in Blood and Blocks Parasite Transmission to Mosquitoes

 , and
, and 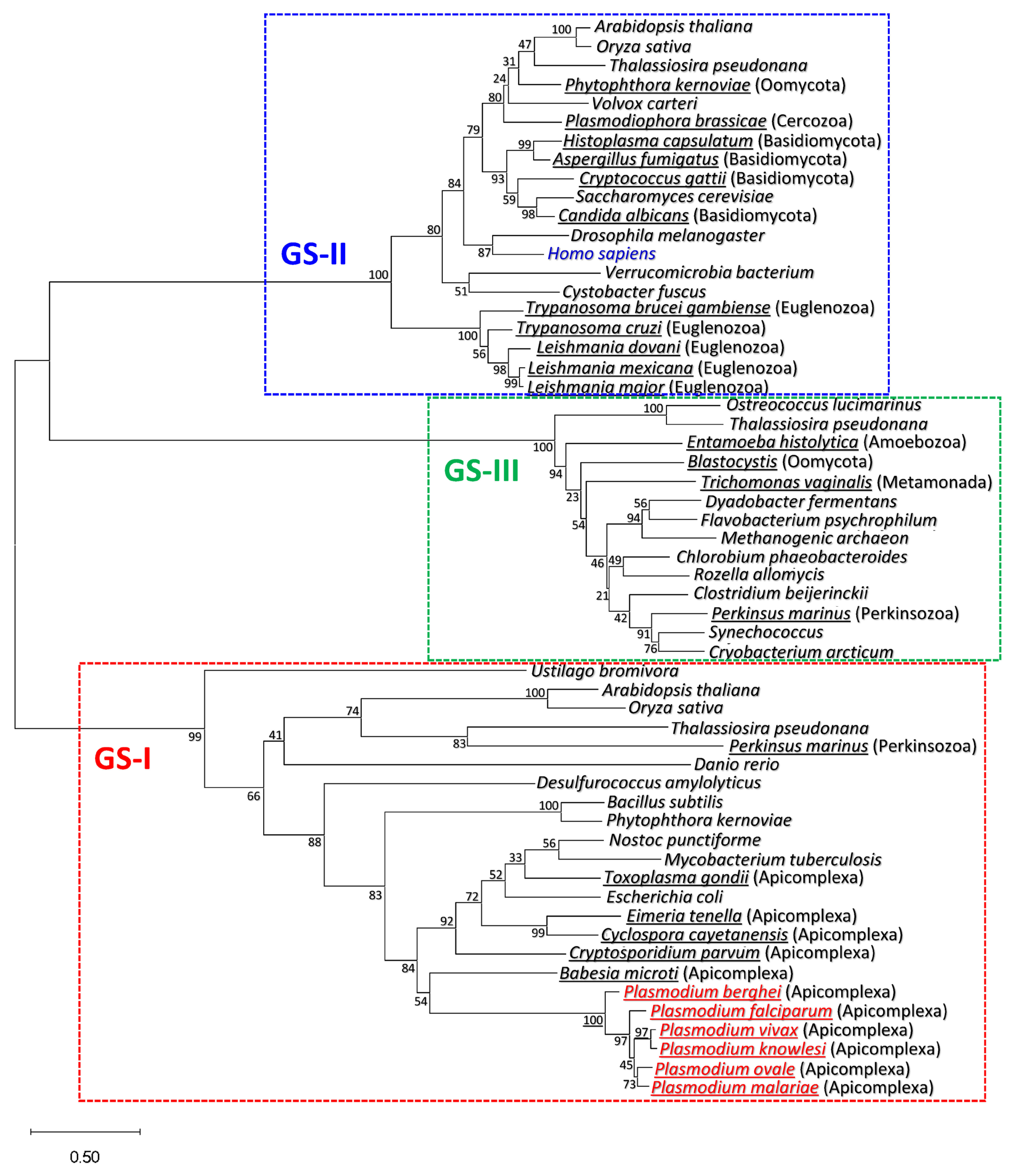{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Mosquitos and Plasmodium Strains
2.3. Human Cell Lines
2.4. Phylogenetic Analysis of Glutamine Synthetase Homologs
2.5. Cloning, Expressing, and Purifying Proteins
2.6. Glutamine Synthetase Assays
2.7. Antimalarial Activity of AST on the Asexual Stage of P. falciparum
2.8. Transmission-Blocking Assays of AST against P. falciparum
2.9. Cytotoxicity Assays
2.10. Membrane Permeability and Stability of AST in Human Cell Lines
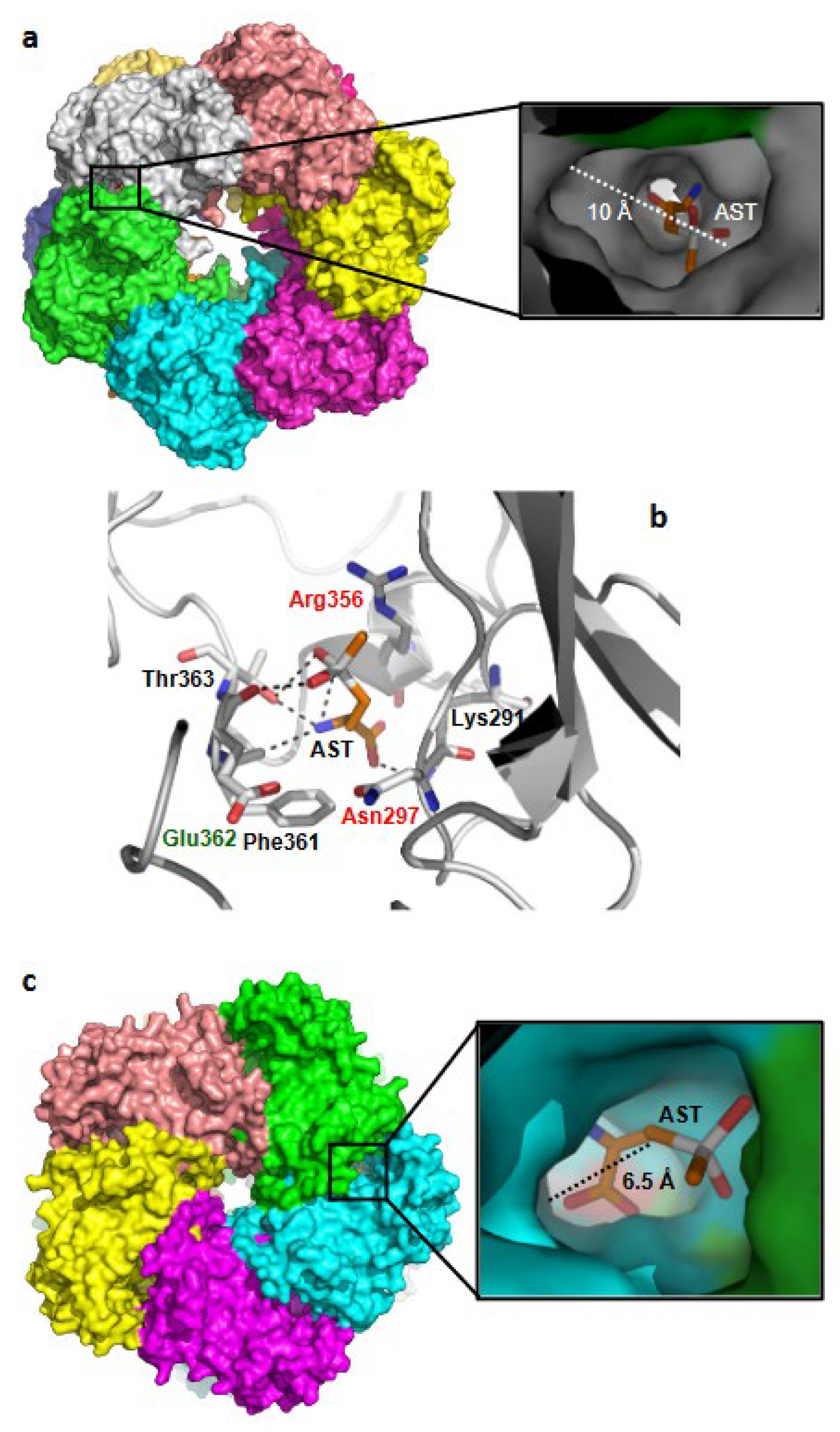
2.11. GS-AST Docking
2.12. Statistics
3. Results
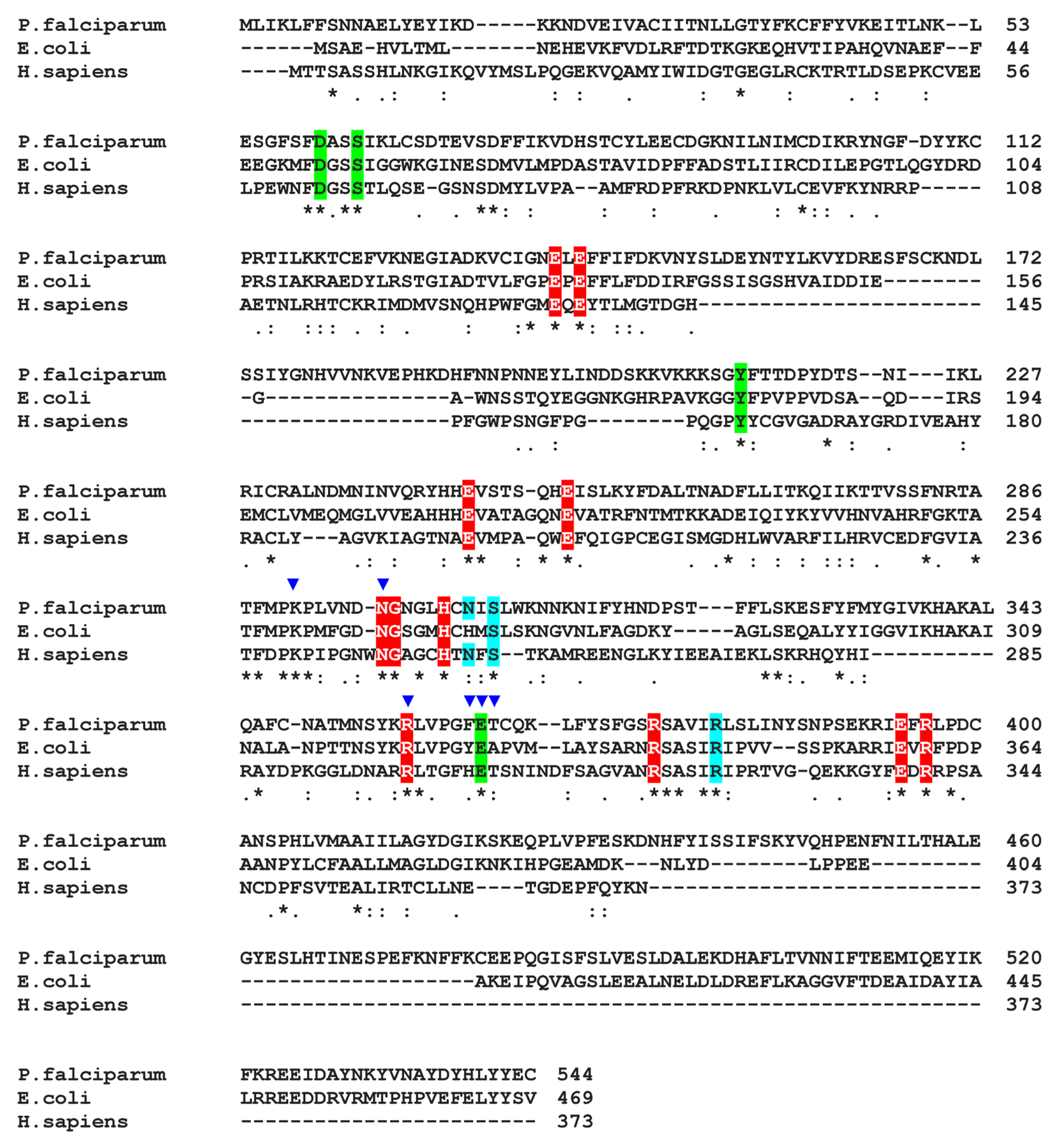
3.1. Plasmodium GS Is Phylogenetically Closer to Prokaryotic GS-I than to Eukaryotic GS-II
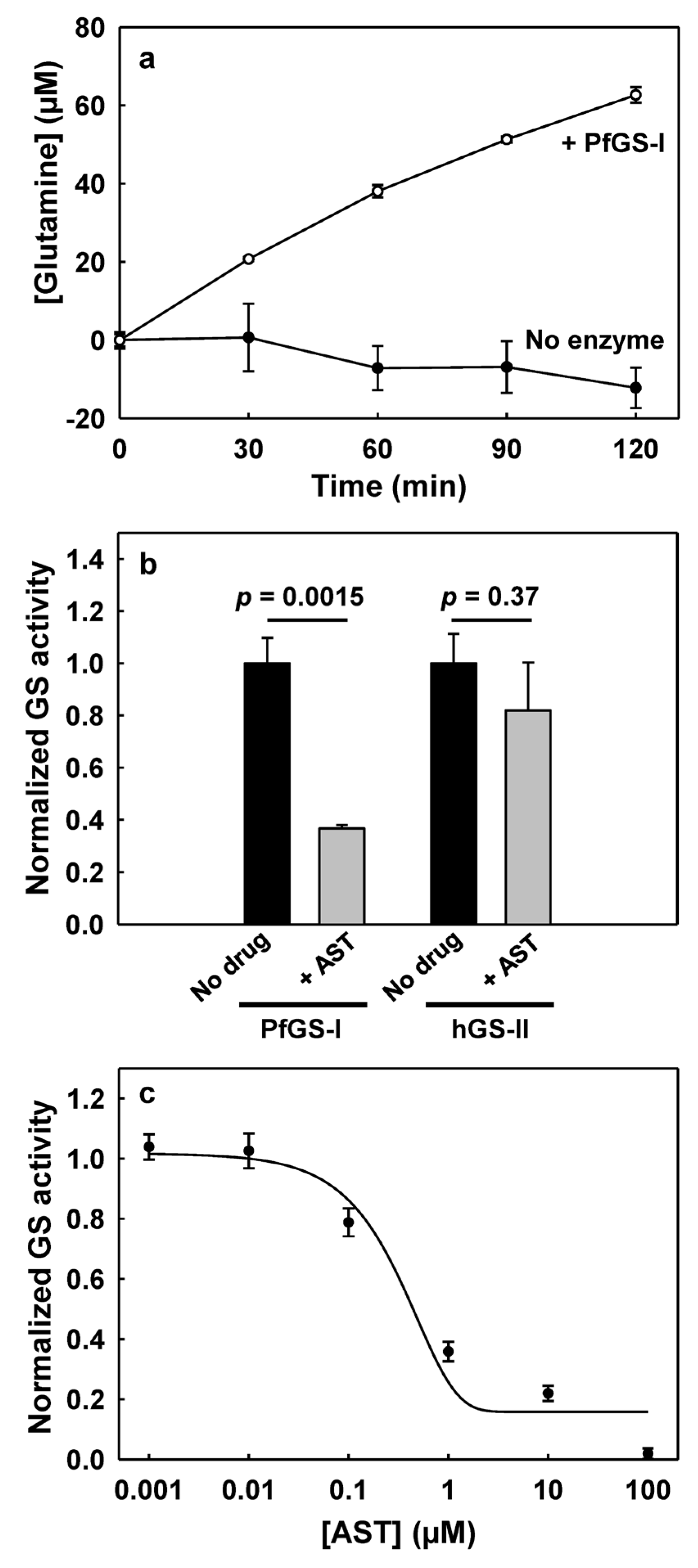
3.2. PfGS-I Is a Functional GS
3.3. AST Effectively Inhibits PfGS-I but Not Human GS-II
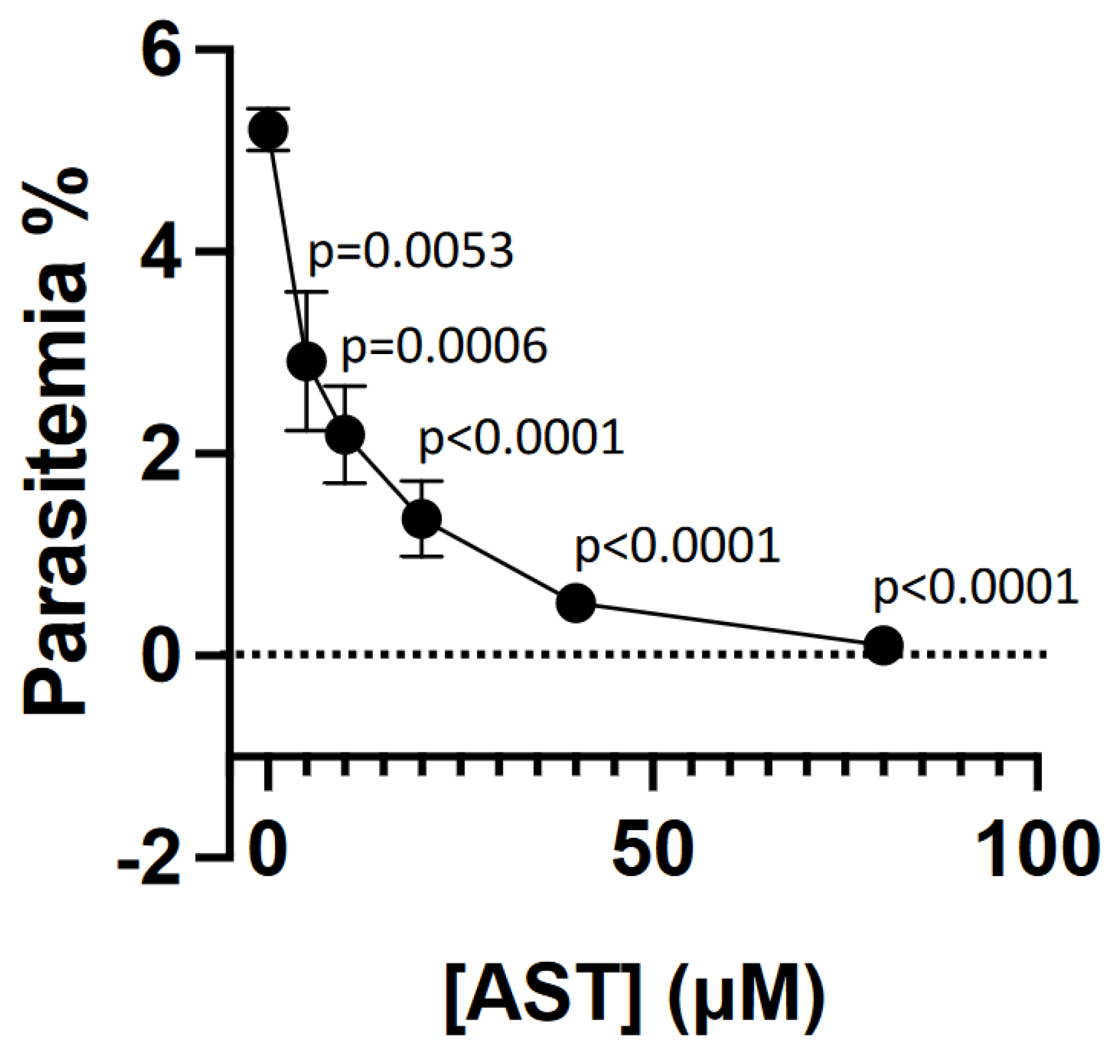
3.4. AST Inhibits Asexual-Stage P. falciparum Proliferation
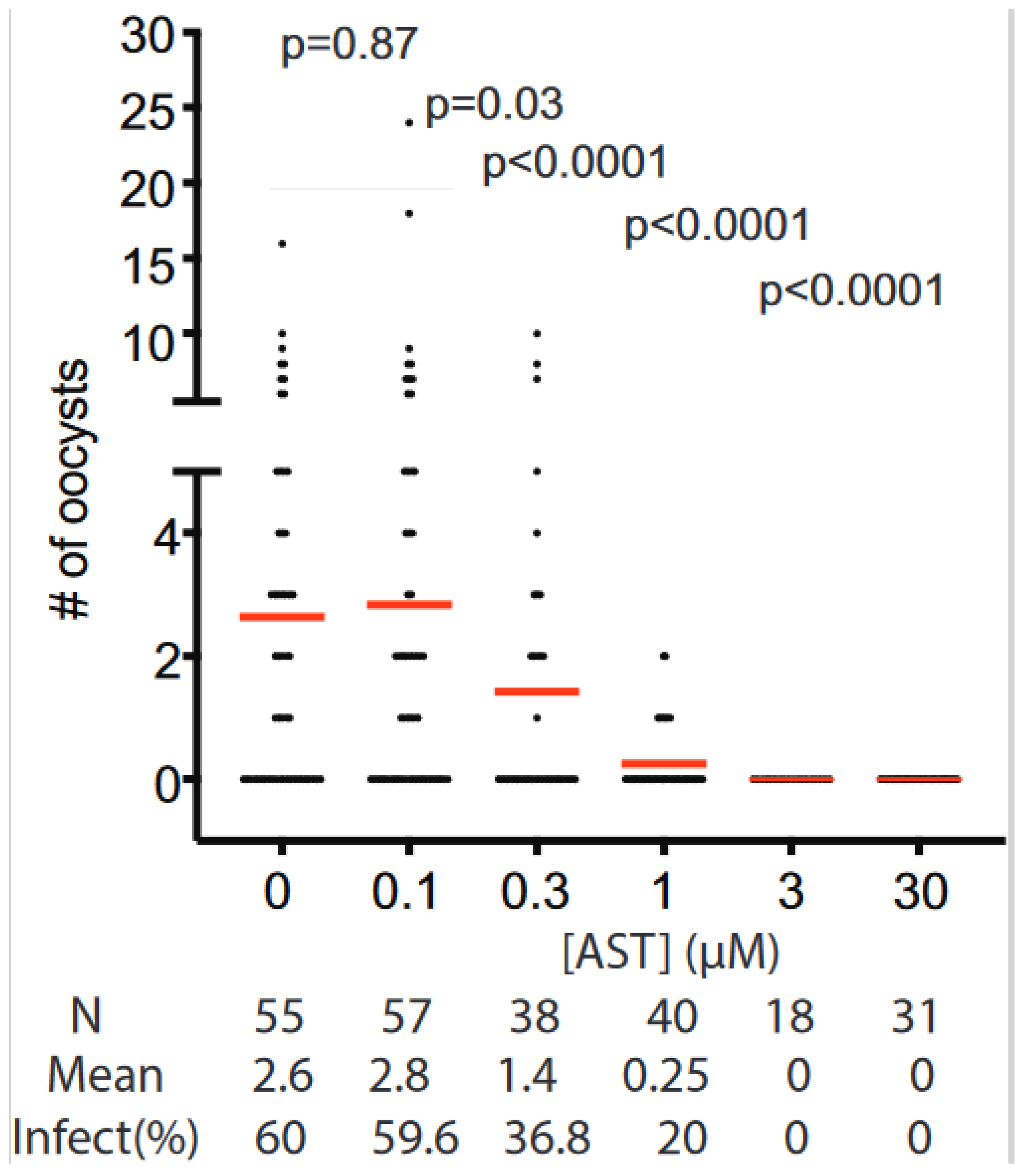
3.5. Transmission Blocking Activity of AST
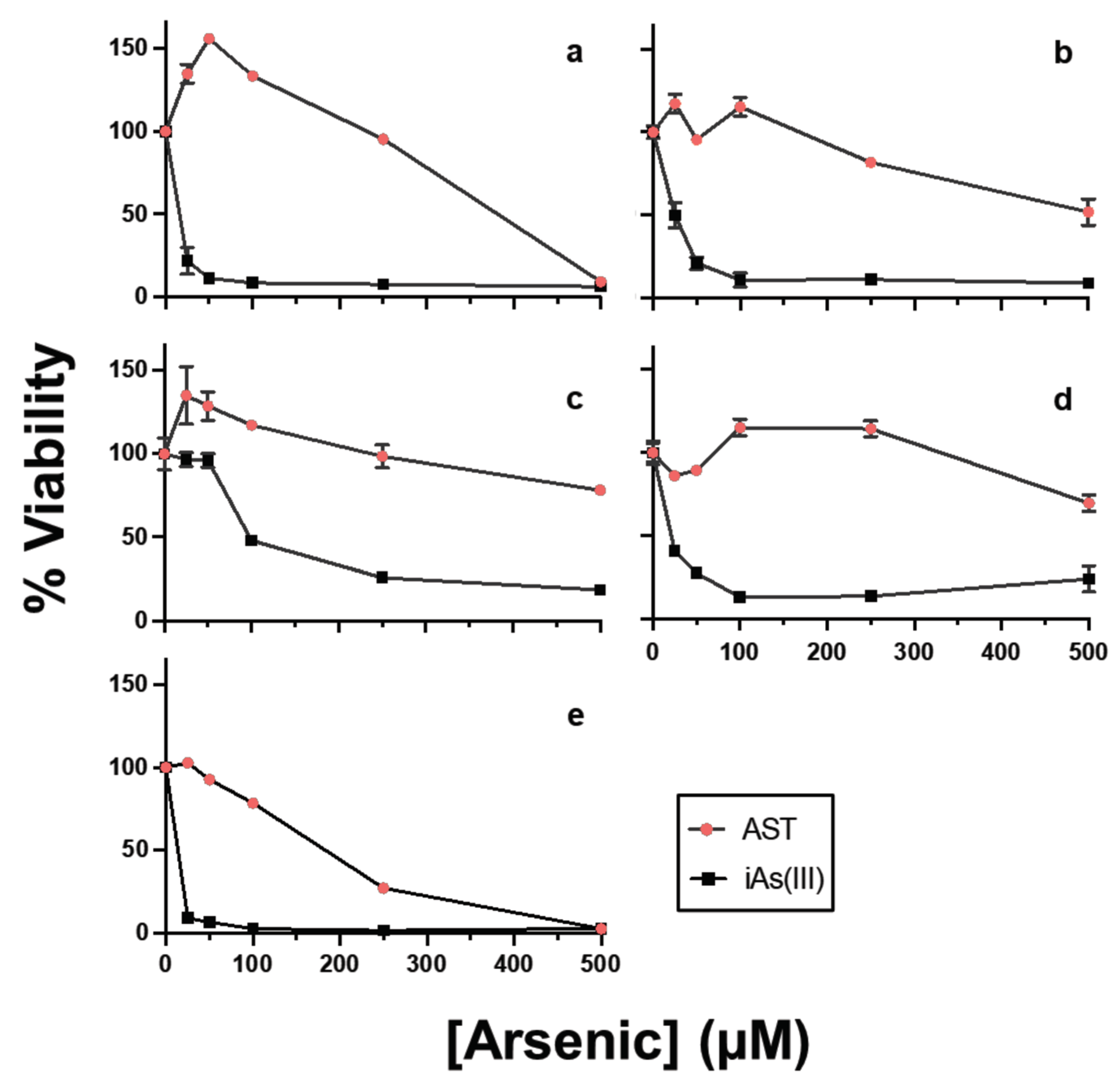
3.6. AST Shows Low Cytotoxicity in Human Cell Lines
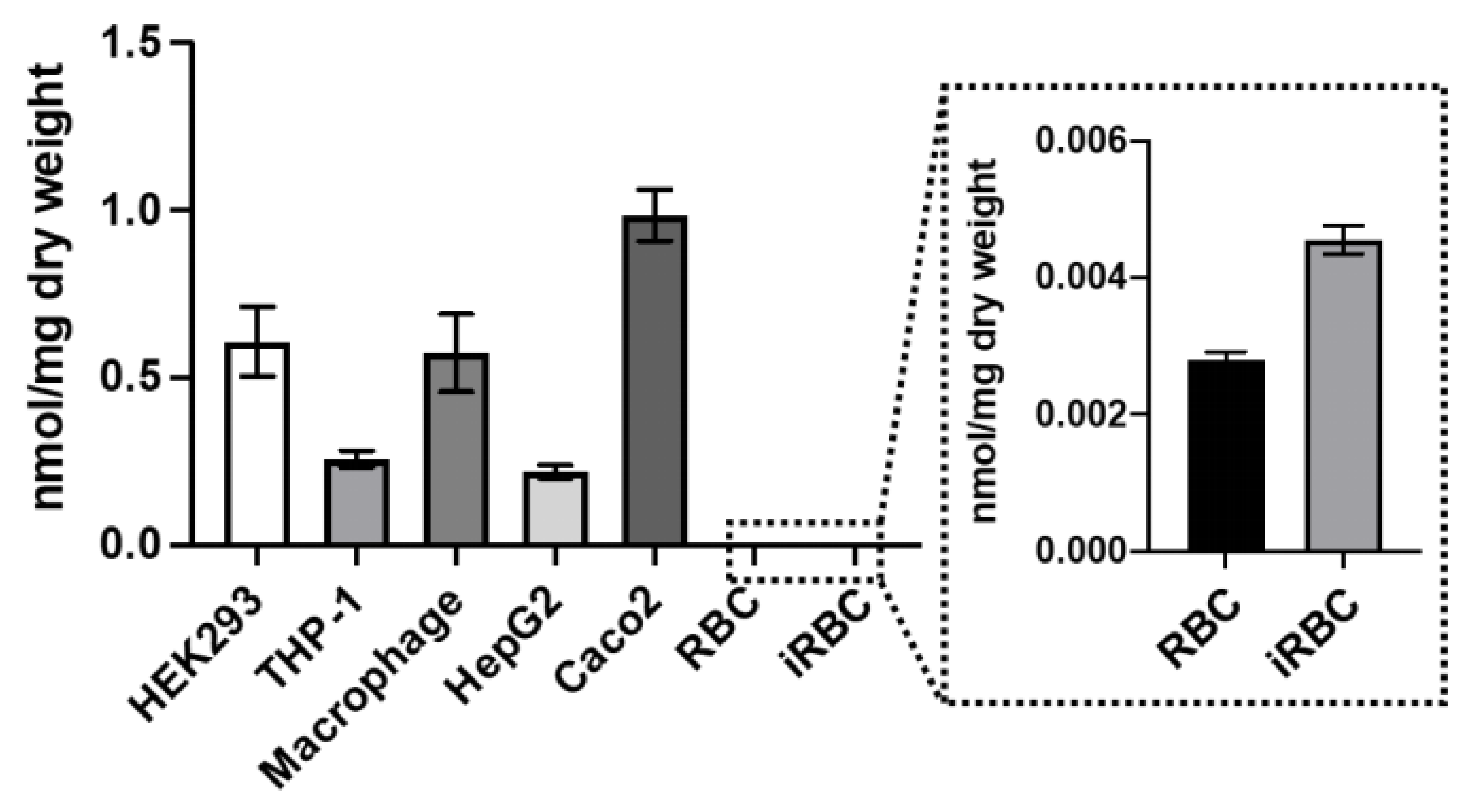
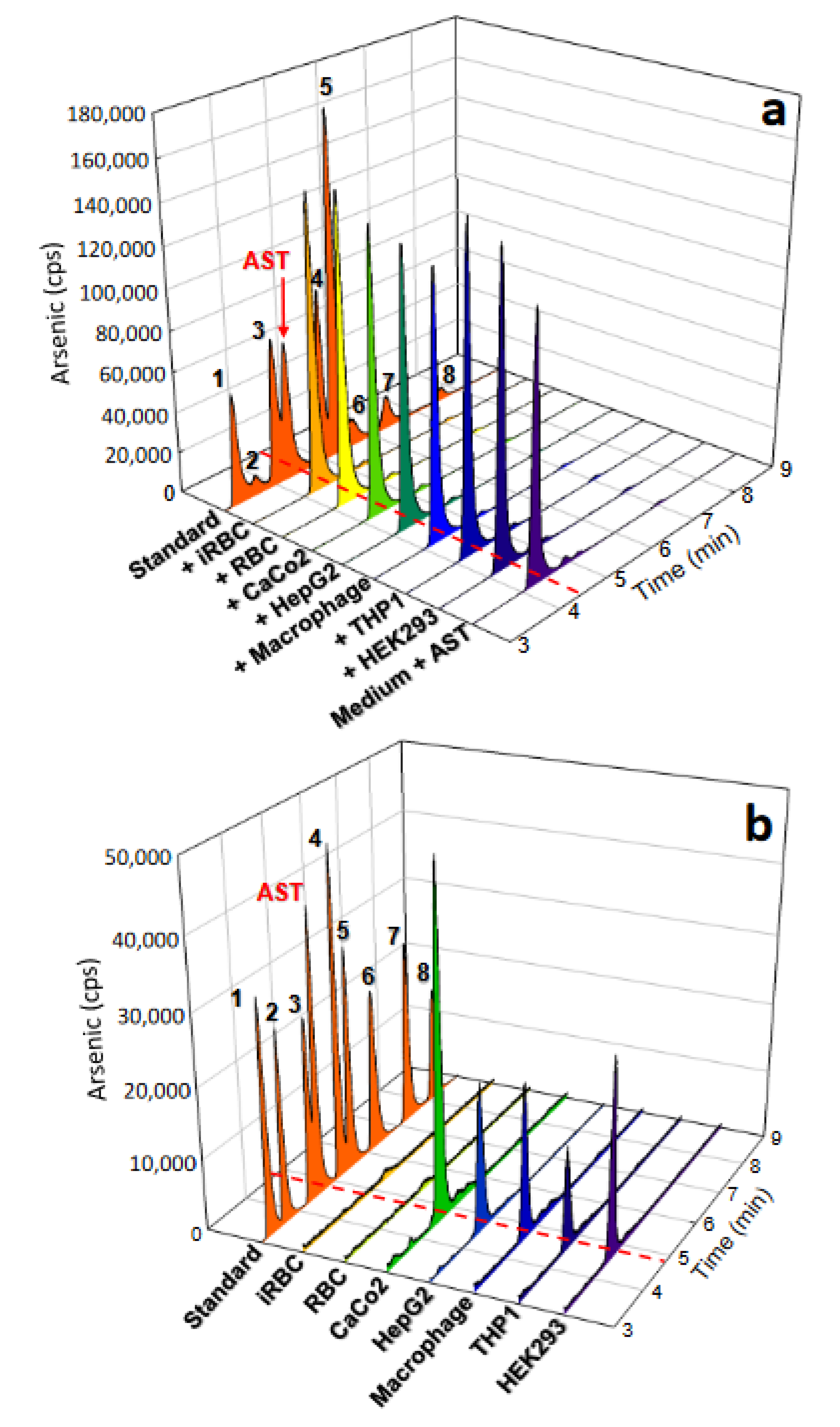
3.7. AST Permeability and Stability in Human Cells
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Monroe, A.; Williams, N.A.; Ogoma, S.; Karema, C.; Okumu, F. Reflections on the 2021 World Malaria Report and the future of malaria control. Malar. J. 2022, 21, 154. [Google Scholar] [CrossRef]
- Nadeem, A.Y.; Shehzad, A.; Islam, S.U.; Al-Suhaimi, E.A.; Lee, Y.S. Mosquirix™ RTS, S/AS01 Vaccine Development, Immunogenicity, and Efficacy. Vaccines 2022, 10, 713. [Google Scholar] [CrossRef] [PubMed]
- Piameu, M.; Nwane, P.; Toussile, W.; Mavridis, K.; Wipf, N.C.; Kouadio, P.F.; Mbakop, L.R.; Mandeng, S.; Ekoko, W.E.; Toto, J.C.; et al. Pyrethroid and Etofenprox Resistance in Anopheles gambiae and Anopheles coluzzii from Vegetable Farms in Yaoundé, Cameroon: Dynamics, Intensity and Molecular Basis. Molecules 2021, 26, 5543. [Google Scholar] [CrossRef] [PubMed]
- Shaw, W.R.; Catteruccia, F. Vector biology meets disease control: Using basic research to fight vector-borne diseases. Nat. Microbiol. 2019, 4, 20–34. [Google Scholar] [CrossRef]
- Chidimatembue, A.; Svigel, S.S.; Mayor, A.; Aíde, P.; Nhama, A.; Nhamussua, L.; Nhacolo, A.; Bassat, Q.; Salvador, C.; Enosse, S.; et al. Molecular surveillance for polymorphisms associated with artemisinin-based combination therapy resistance in Plasmodium falciparum isolates collected in Mozambique, 2018. Malar. J. 2021, 20, 398. [Google Scholar] [CrossRef]
- Balikagala, B.; Fukuda, N.; Ikeda, M.; Katuro, O.T.; Tachibana, S.I.; Yamauchi, M.; Opio, W.; Emoto, S.; Anywar, D.A.; Kimura, E.; et al. Evidence of Artemisinin-Resistant Malaria in Africa. N. Engl. J. Med. 2021, 385, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Paton, D.G.; Childs, L.M.; Itoe, M.A.; Holmdahl, I.E.; Buckee, C.O.; Catteruccia, F. Exposing Anopheles mosquitoes to antimalarials blocks Plasmodium parasite transmission. Nature 2019, 567, 239–243. [Google Scholar] [CrossRef]
- Mojab, F. Antimalarial natural products: A review. Avicenna. J. Phytomed. 2012, 2, 52–62. [Google Scholar]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef]
- Niu, G.; Annamalai, T.; Wang, X.; Li, S.; Munga, S.; Niu, G.; Tse-Dinh, Y.C.; Li, J. A diverse global fungal library for drug discovery. PeerJ 2020, 8, e10392. [Google Scholar] [CrossRef]
- Niu, G.; Hao, Y.; Wang, X.; Gao, J.M.; Li, J. Fungal Metabolite Asperaculane B Inhibits Malaria Infection and Transmission. Molecules 2020, 25, 3018. [Google Scholar] [CrossRef] [PubMed]
- Niu, G.; Wang, B.; Zhang, G.; King, J.B.; Cichewicz, R.H.; Li, J. Targeting mosquito FREP1 with a fungal metabolite blocks malaria transmission. Sci. Rep. 2015, 5, 14694. [Google Scholar] [CrossRef] [PubMed]
- Niu, G.; Wang, X.; Hao, Y.; Kandel, S.; Niu, G.; Raptis, R.G.; Li, J. A novel fungal metabolite inhibits Plasmodium falciparum transmission and infection. Parasit. Vectors 2021, 14, 177. [Google Scholar] [CrossRef] [PubMed]
- Niu, G.; Kalani, K.; Wang, X.; Li, J. Sterigmatocystin Limits Plasmodium falciparum Proliferation and Transmission. Pharmaceuticals 2021, 14, 1238. [Google Scholar] [CrossRef] [PubMed]
- Wadi, I.; Nath, M.; Anvikar, A.R.; Singh, P.; Sinha, A. Recent advances in transmission-blocking drugs for malaria elimination. Future Med. Chem. 2019, 11, 3047–3088. [Google Scholar] [CrossRef]
- Kuramata, M.; Sakakibara, F.; Kataoka, R.; Yamazaki, K.; Baba, K.; Ishizaka, M.; Hiradate, S.; Kamo, T.; Ishikawa, S. Arsinothricin, a novel organoarsenic species produced by a rice rhizosphere bacterium. Environ. Chem. 2016, 13, 723. [Google Scholar] [CrossRef]
- Nadar, V.S.; Chen, J.; Dheeman, D.S.; Galván, A.E.; Yoshinaga-Sakurai, K.; Kandavelu, P.; Sankaran, B.; Kuramata, M.; Ishikawa, S.; Rosen, B.P.; et al. Arsinothricin, an arsenic-containing non-proteinogenic amino acid analog of glutamate, is a broad-spectrum antibiotic. Commun. Biol. 2019, 2, 131. [Google Scholar] [CrossRef] [PubMed]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Paul, N.P.; Galván, A.E.; Yoshinaga-Sakurai, K.; Rosen, B.P.; Yoshinaga, M. Arsenic in medicine: Past, present and future. Biometals 2022, 36, 283–301. [Google Scholar] [CrossRef] [PubMed]
- Kritharis, A.; Bradley, T.P.; Budman, D.R. The evolving use of arsenic in pharmacotherapy of malignant disease. Ann. Hematol. 2013, 92, 719–730. [Google Scholar] [CrossRef]
- Brun, R.; Blum, J.; Chappuis, F.; Burri, C. Human African trypanosomiasis. Lancet 2010, 375, 148–159. [Google Scholar] [CrossRef]
- Welch, B.; Smit, E.; Cardenas, A.; Hystad, P.; Kile, M.L. Trends in urinary arsenic among the U.S. population by drinking water source: Results from the National Health and Nutritional Examinations Survey 2003–2014. Environ. Res. 2018, 162, 8–17. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, S.J. Poisoning the Devil. Cell 2017, 168, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Galván, A.E.; Paul, N.P.; Chen, J.; Yoshinaga-Sakurai, K.; Utturkar, S.M.; Rosen, B.P.; Yoshinaga, M. Identification of the Biosynthetic Gene Cluster for the Organoarsenical Antibiotic Arsinothricin. Microbiol. Spectr. 2021, 9, e0050221. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.L. Simultaneous determination of arsenate and phosphate in natural waters. Environ. Sci. Technol. 1971, 5, 411–414. [Google Scholar] [CrossRef]
- Suzol, S.H.; Hasan Howlader, A.; Galván, A.E.; Radhakrishnan, M.; Wnuk, S.F.; Rosen, B.P.; Yoshinaga, M. Semisynthesis of the Organoarsenical Antibiotic Arsinothricin. J. Nat. Prod. 2020, 83, 2809–2813. [Google Scholar] [CrossRef] [PubMed]
- Naranmandura, H.; Suzuki, N.; Iwata, K.; Hirano, S.; Suzuki, K.T. Arsenic metabolism and thioarsenicals in hamsters and rats. Chem. Res. Toxicol. 2007, 20, 616–624. [Google Scholar] [CrossRef]
- Naranmandura, H.; Suzuki, N.; Suzuki, K.T. Trivalent arsenicals are bound to proteins during reductive methylation. Chem. Res. Toxicol. 2006, 19, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, X.; Zhang, G.; Githure, J.I.; Yan, G.; James, A.A. Genome-block expression-assisted association studies discover malaria resistance genes in Anopheles gambiae. Proc. Natl. Acad. Sci. USA 2013, 110, 20675–20680. [Google Scholar] [CrossRef]
- Zhang, G.; Niu, G.; Franca, C.M.; Dong, Y.; Wang, X.; Butler, N.S.; Dimopoulos, G.; Li, J. Anopheles Midgut FREP1 Mediates Plasmodium Invasion. J. Biol. Chem. 2015, 290, 16490–16501. [Google Scholar] [CrossRef]
- McCartan, S.; Powell, L.A.; Green, B.D.; McEneny, J.; McGinty, A. The anti-atherogenic effects of eicosapentaenoic and docosahexaenoic acid are dependent on the stage of THP-1 macrophage differentiation. J. Funct. Foods 2015, 19, 958–969. [Google Scholar] [CrossRef]
- de Carvalho Fernandes, G.; Turchetto-Zolet, A.C.; Pereira Passaglia, L.M. Glutamine synthetase evolutionary history revisited: Tracing back beyond the Last Universal Common Ancestor. Evolution 2022, 76, 605–622. [Google Scholar] [CrossRef]
- Rueckert, S.; Pipaliya, S.V.; Dacks, J.B. Evolution: Parallel Paths to Parasitism in the Apicomplexa. Curr. Biol. 2019, 29, R836–R839. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Hall, B.G. Building phylogenetic trees from molecular data with MEGA. Mol. Biol. Evol. 2013, 30, 1229–1235. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic. Acids. Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Twentyman, P.R.; Luscombe, M. A study of some variables in a tetrazolium dye (MTT) based assay for cell growth and chemosensitivity. Br. J. Cancer 1987, 56, 279–285. [Google Scholar] [CrossRef]
- Ho, C.M.; Li, X.; Lai, M.; Terwilliger, T.C.; Beck, J.R.; Wohlschlegel, J.; Goldberg, D.E.; Fitzpatrick, A.W.P.; Zhou, Z.H. Bottom-up structural proteomics: CryoEM of protein complexes enriched from the cellular milieu. Nat. Methods 2020, 17, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Krajewski, W.W.; Collins, R.; Holmberg-Schiavone, L.; Jones, T.A.; Karlberg, T.; Mowbray, S.L. Crystal structures of mammalian glutamine synthetases illustrate substrate-induced conformational changes and provide opportunities for drug and herbicide design. J. Mol. Biol. 2008, 375, 217–228. [Google Scholar] [CrossRef]
- Aurrecoechea, C.; Brestelli, J.; Brunk, B.P.; Dommer, J.; Fischer, S.; Gajria, B.; Gao, X.; Gingle, A.; Grant, G.; Harb, O.S.; et al. PlasmoDB: A functional genomic database for malaria parasites. Nucleic. Acids Res. 2009, 37, D539-43. [Google Scholar] [CrossRef] [PubMed]
- Grassi, F.; Moretto, N.; Rivetti, C.; Cellai, S.; Betti, M.; Márquez, A.J.; Maraini, G.; Ottonello, S. Structural and functional properties of lengsin, a pseudo-glutamine synthetase in the transparent human lens. Biochem. Biophys Res. Commun. 2006, 350, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Díaz, E.; Yerbanga, R.S.; Lefèvre, T.; Cohuet, A.; Rowley, M.J.; Ouedraogo, J.B.; Corces, V.G. Epigenetic regulation of Plasmodium falciparum clonally variant gene expression during development in Anopheles gambiae. Sci. Rep. 2017, 7, 40655. [Google Scholar] [CrossRef]
- López-Barragán, M.J.; Lemieux, J.; Quiñones, M.; Williamson, K.C.; Molina-Cruz, A.; Cui, K.; Barillas-Mury, C.; Zhao, K.; Su, X.Z. Directional gene expression and antisense transcripts in sexual and asexual stages of Plasmodium falciparum. BMC Genom. 2011, 12, 587. [Google Scholar] [CrossRef] [PubMed]
- Le Roch, K.G.; Zhou, Y.; Blair, P.L.; Grainger, M.; Moch, J.K.; Haynes, J.D.; De La Vega, P.; Holder, A.A.; Batalov, S.; Carucci, D.J.; et al. Discovery of gene function by expression profiling of the malaria parasite life cycle. Science 2003, 301, 1503–1508. [Google Scholar] [CrossRef] [PubMed]
- Counihan, N.A.; Modak, J.K.; de Koning-Ward, T.F. How Malaria Parasites Acquire Nutrients From Their Host. Front. Cell. Dev. Biol. 2021, 9, 649184. [Google Scholar] [CrossRef]
- Boyle, M.J.; Wilson, D.W.; Richards, J.S.; Riglar, D.T.; Tetteh, K.K.; Conway, D.J.; Ralph, S.A.; Baum, J.; Beeson, J.G. Isolation of viable Plasmodium falciparum merozoites to define erythrocyte invasion events and advance vaccine and drug development. Proc. Natl. Acad. Sci. USA 2010, 107, 14378–14383. [Google Scholar] [CrossRef]
- Kosikowska, P.; Bochno, M.; Macegoniuk, K.; Forlani, G.; Kafarski, P.; Berlicki, Ł. Bisphosphonic acids as effective inhibitors of Mycobacterium tuberculosis glutamine synthetase. J. Enzyme Inhib Med. Chem. 2016, 31, 931–938. [Google Scholar] [CrossRef]
- Opperdoes, F.R.; Coombs, G.H. Metabolism of Leishmania: Proven and predicted. Trends Parasitol. 2007, 23, 149–158. [Google Scholar] [CrossRef]
- Saunders, E.C.; Ng, W.W.; Kloehn, J.; Chambers, J.M.; Ng, M.; McConville, M.J. Induction of a stringent metabolic response in intracellular stages of Leishmania mexicana leads to increased dependence on mitochondrial metabolism. PLoS Pathog. 2014, 10, e1003888. [Google Scholar] [CrossRef]
- Magdaleno, A.; Suárez Mantilla, B.; Rocha, S.C.; Pral, E.M.; Silber, A.M. The Involvement of Glutamate Metabolism in the Resistance to Thermal, Nutritional, and Oxidative Stress in Trypanosoma cruzi. Enzyme. Res. 2011, 2011, 486928. [Google Scholar] [CrossRef]
- Kumar, V.; Ghosh, S.; Roy, K.; Pal, C.; Singh, S. Deletion of Glutamine Synthetase Gene Disrupts the Survivability and Infectivity of Leishmania donovani. Front. Cell. Infect. Microbiol. 2021, 11, 622266. [Google Scholar] [CrossRef] [PubMed]
- Crispim, M.; Damasceno, F.S.; Hernández, A.; Barisón, M.J.; Pretto Sauter, I.; Souza Pavani, R.; Santos Moura, A.; Pral, E.M.F.; Cortez, M.; Elias, M.C.; et al. The glutamine synthetase of Trypanosoma cruzi is required for its resistance to ammonium accumulation and evasion of the parasitophorous vacuole during host-cell infection. PLoS Negl. Trop. Dis. 2018, 12, e0006170. [Google Scholar] [CrossRef] [PubMed]
- van Schalkwyk, D.A.; Chan, X.W.; Misiano, P.; Gagliardi, S.; Farina, C.; Saliba, K.J. Inhibition of Plasmodium falciparum pH regulation by small molecule indole derivatives results in rapid parasite death. Biochem. Pharmacol. 2010, 79, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Harb, O.S.; Roos, D.S. ToxoDB: Functional Genomics Resource for Toxoplasma and Related Organisms. Methods Mol. Biol. 2020, 2071, 27–47. [Google Scholar] [PubMed]
- Guiton, P.S.; Sagawa, J.M.; Fritz, H.M.; Boothroyd, J.C. An in vitro model of intestinal infection reveals a developmentally regulated transcriptome of Toxoplasma sporozoites and a NF-κB-like signature in infected host cells. PLoS ONE 2017, 12, e0173018. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, C.; Maier, S.; Walker, R.A.; Rehrauer, H.; Joekel, D.E.; Winiger, R.R.; Basso, W.U.; Grigg, M.E.; Hehl, A.B.; Deplazes, P.; et al. An experimental genetically attenuated live vaccine to prevent transmission of Toxoplasma gondii by cats. Sci. Rep. 2019, 9, 1474. [Google Scholar] [CrossRef] [PubMed]
- Fritz, H.M.; Buchholz, K.R.; Chen, X.; Durbin-Johnson, B.; Rocke, D.M.; Conrad, P.A.; Boothroyd, J.C. Transcriptomic analysis of toxoplasma development reveals many novel functions and structures specific to sporozoites and oocysts. PLoS ONE 2012, 7, e29998. [Google Scholar] [CrossRef]
- Behnke, M.S.; Zhang, T.P.; Dubey, J.P.; Sibley, L.D. Toxoplasma gondii merozoite gene expression analysis with comparison to the life cycle discloses a unique expression state during enteric development. BMC Genom. 2014, 15, 350. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.A.; Sharman, P.A.; Miller, C.M.; Lippuner, C.; Okoniewski, M.; Eichenberger, R.M.; Ramakrishnan, C.; Brossier, F.; Deplazes, P.; Hehl, A.B.; et al. RNA Seq analysis of the Eimeria tenella gametocyte transcriptome reveals clues about the molecular basis for sexual reproduction and oocyst biogenesis. BMC Genom. 2015, 16, 94. [Google Scholar] [CrossRef] [PubMed]
- Reid, A.J.; Blake, D.P.; Ansari, H.R.; Billington, K.; Browne, H.P.; Bryant, J.; Dunn, M.; Hung, S.S.; Kawahara, F.; Miranda-Saavedra, D.; et al. Genomic analysis of the causative agents of coccidiosis in domestic chickens. Genome. Res. 2014, 24, 1676–1685. [Google Scholar] [CrossRef]
- Ehret, T.; Spork, S.; Dieterich, C.; Lucius, R.; Heitlinger, E. Dual RNA-seq reveals no plastic transcriptional response of the coccidian parasite Eimeria falciformis to host immune defenses. BMC Genom. 2017, 18, 686. [Google Scholar] [CrossRef] [PubMed]
- Heiges, M.; Wang, H.; Robinson, E.; Aurrecoechea, C.; Gao, X.; Kaluskar, N.; Rhodes, P.; Wang, S.; He, C.Z.; Su, Y.; et al. CryptoDB: A Cryptosporidium bioinformatics resource update. Nucleic. Acids Res. 2006, 34, D419-22. [Google Scholar] [CrossRef] [PubMed]
- Tandel, J.; English, E.D.; Sateriale, A.; Gullicksrud, J.A.; Beiting, D.P.; Sullivan, M.C.; Pinkston, B.; Striepen, B. Life cycle progression and sexual development of the apicomplexan parasite Cryptosporidium parvum. Nat. Microbiol. 2019, 4, 2226–2236. [Google Scholar] [CrossRef]
- Chaudhary, K.; Roos, D.S. Protozoan genomics for drug discovery. Nat. Biotechnol. 2005, 23, 1089–1091. [Google Scholar] [CrossRef]
- Seeber, F.; Limenitakis, J.; Soldati-Favre, D. Apicomplexan mitochondrial metabolism: A story of gains, losses and retentions. Trends Parasitol. 2008, 24, 468–478. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, C.; Otto, T.D.; Oberstaller, J.; Liao, X.; Adapa, S.R.; Udenze, K.; Bronner, I.F.; Casandra, D.; Mayho, M.; et al. Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science 2018, 360, eaap7847. [Google Scholar] [CrossRef]
- Mensa-Wilmot, K. How Physiologic Targets Can Be Distinguished from Drug-Binding Proteins. Mol. Pharmacol. 2021, 100, 1–6. [Google Scholar] [CrossRef]
- Yang, J.; He, Y.; Li, Y.; Zhang, X.; Wong, Y.K.; Shen, S.; Zhong, T.; Zhang, J.; Liu, Q.; Wang, J. Advances in the research on the targets of anti-malaria actions of artemisinin. Pharmacol. Ther. 2020, 216, 107697. [Google Scholar] [CrossRef] [PubMed]
- Bennink, S.; Kiesow, M.J.; Pradel, G. The development of malaria parasites in the mosquito midgut. Cell. Microbiol. 2016, 18, 905–918. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoshinaga, M.; Niu, G.; Yoshinaga-Sakurai, K.; Nadar, V.S.; Wang, X.; Rosen, B.P.; Li, J. Arsinothricin Inhibits Plasmodium falciparum Proliferation in Blood and Blocks Parasite Transmission to Mosquitoes. Microorganisms 2023, 11, 1195. https://doi.org/10.3390/microorganisms11051195
Yoshinaga M, Niu G, Yoshinaga-Sakurai K, Nadar VS, Wang X, Rosen BP, Li J. Arsinothricin Inhibits Plasmodium falciparum Proliferation in Blood and Blocks Parasite Transmission to Mosquitoes. Microorganisms. 2023; 11(5):1195. https://doi.org/10.3390/microorganisms11051195
Chicago/Turabian StyleYoshinaga, Masafumi, Guodong Niu, Kunie Yoshinaga-Sakurai, Venkadesh S. Nadar, Xiaohong Wang, Barry P. Rosen, and Jun Li. 2023. "Arsinothricin Inhibits Plasmodium falciparum Proliferation in Blood and Blocks Parasite Transmission to Mosquitoes" Microorganisms 11, no. 5: 1195. https://doi.org/10.3390/microorganisms11051195